UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |||||
For the fiscal year ended December 31 , 2025
or
| TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |||||
For the transition period from to
Commission file number: 001-33137

(Exact Name of Registrant as Specified in Its Charter)
| (State or Other Jurisdiction of Incorporation or Organization) | (I.R.S. Employer Identification No.) | |||||||
| (Address of Principal Executive Offices) | ||||||||
| (City) | (State) | (Zip Code) | ||||||
Registrant's Telephone Number, Including Area Code: (240 ) 631-3200
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Trading Symbol(s) | Name of Each Exchange on Which Registered | ||||||
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of Securities Act. Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of "large accelerated filer," "accelerated filer," "non-accelerated filer", "smaller reporting company" and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ☐ Accelerated filer ☒ Non-accelerated filer ☐ Smaller reporting company ☐ Emerging growth company ☐
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management's assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. Yes ☒ No ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant's executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of voting and non-voting common equity held by non-affiliates of the registrant as of June 30, 2025 was approximately $342.7 million based on the price at which the registrant's common stock was last sold on that date as reported on the New York Stock Exchange.
As of February 19, 2026, the registrant had 51,770,857 shares of common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant's definitive proxy statement for its 2026 annual meeting of stockholders, which is expected to be filed with the United States Securities and Exchange Commission not later than 120 days after the end of the registrant's fiscal year ended December 31, 2025, are incorporated by reference into Part III of this Annual Report on Form 10-K. With the exception of the portions of the registrant's definitive proxy statement for its 2026 annual meeting of stockholders that are expressly incorporated by reference into this Annual Report on Form 10-K, such proxy statement shall not be deemed filed as part of this Annual Report on Form 10-K.
EMERGENT BIOSOLUTIONS INC. AND SUBSIDIARIES
Annual Report on Form 10-K
Fiscal Year Ended December 31, 2025
TABLE OF CONTENTS
| Page | ||||||||
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K and the documents we incorporate by reference include forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. All statements, other than statements of historical fact, including statements regarding the future performance of Emergent BioSolutions Inc. or any of our businesses, our business strategy, future operations, future financial position, future revenues and earnings, our ability to achieve the objectives of our restructuring initiatives, acquisitions and divestitures, including our future results, projected costs, prospects, plans and objectives of management, are forward-looking statements. We generally identify forward-looking statements by using words like “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “forecast,” “future,” “goal,” “intend,” “may,” “plan,” “position,” “possible,” “potential,” “predict,” “project,” “should,” “target,” “will,” “would,” and similar expressions or variations thereof, or the negative thereof, but these terms are not the exclusive means of identifying such statements. These forward-looking statements are based on our current intentions, beliefs, assumptions and expectations regarding future events based on information that is currently available. You should realize that if underlying assumptions prove inaccurate or unknown risks or uncertainties materialize, actual results could differ materially from our expectations. You are, therefore, cautioned not to place undue reliance on any forward-looking statement contained herein. Any such forward-looking statement speaks only as of the date on which such statement is made and, except as required by law, we do not undertake any obligation to update any forward-looking statement to reflect new information, events or circumstances.
There are a number of important factors that could cause our actual results to differ materially from those indicated by such forward-looking statements, including, among others:
•the availability of U.S. government (“USG”) funding for contracts related to procurement of our medical countermeasures (“MCM”) products, including CYFENDUS® (Anthrax Vaccine Adsorbed (AVA), Adjuvanted), ACAM2000® (Smallpox (Vaccinia) Vaccine, Live), CNJ-016® (Vaccinia Immune Globulin Intravenous, Human (VIGIV)), BAT® (Botulism Antitoxin Heptavalent (A,B,C,D,E,F,G)-(Equine)), BioThrax® (Anthrax Vaccine Adsorbed), EbangaTM (ansuvimab-zykl) and/or TEMBEXA® among others, as well as contracts related to development of medical countermeasures;
•our ability to meet our commitments to quality and compliance in all of our manufacturing operations;
•our ability to negotiate additional USG procurement or follow-on contracts for our MCM products that have expired or will be expiring;
•the commercial availability and impact of a generic and competitive marketplace on future sales of NARCAN® (naloxone HCL) Nasal Spray and over-the-counter NARCAN® Nasal Spray and KLOXXADO® Nasal Spray;
•our ability to perform under our contracts with the USG, including the timing of and specifications relating to deliveries;
•the ability of our contractors and suppliers to maintain compliance with current good manufacturing practices and other regulatory obligations;
•our ability to collect reimbursement for raw materials and payment of service fees from our Bioservices customers;
•the results of pending government investigations and their potential impact on our business;
•our ability to satisfy the conditions of our litigation settlement agreements, and the potential impact of such agreements, including the funds to resolve related litigation, on our business;
•our ability to comply with the operating and financial covenants required by (i) our term loan facility (the “Term Loan Facility”) under a credit agreement, dated August 30, 2024, among the Company, the lenders from time to time party thereto and OHA Agency LLC, as administrative agent, (ii) our revolving credit facility (the “Revolving Credit Facility”, and together with the Term Loan Facility, the “Senior Secured Credit Facilities”) under a credit agreement, dated September 30, 2024, among the Company, certain subsidiary borrowers, the lenders from time to time party thereto and Wells Fargo, National Association, as Agent, and (iii) our 3.875% Senior Unsecured Notes due 2028 (the “Senior Unsecured Notes”);
•our ability to maintain adequate internal control over financial reporting and to prepare accurate financial statements in a timely manner;
•our ability to maintain sufficient cash flow from our operations to pay our substantial debt, both now and in the future;
•our ability to invest in our business operations as a result of our current indebtedness;
•the impact of our share and debt repurchase programs;
1
•the procurement of our product candidates by USG entities under regulatory authorities that permit government procurement of certain medical products prior to United States ("U.S.") Food and Drug Administration (“FDA”) marketing authorization, and corresponding procurement by government entities outside the United States;
•the success of our commercialization, marketing and manufacturing capabilities and strategy;
•our ability to identify and acquire companies, businesses, products or product candidates that satisfy our selection criteria;
•our ability to attract and retain qualified personnel;
•our ability to adequately secure and protect our intellectual property rights;
•the impact of cybersecurity incidents, including the risks from the unauthorized access, interruption, failure or compromise of our information systems or those of our business partners, collaborators or other third parties; and
•the accuracy of our estimates regarding future revenues, expenses, capital requirements and need for additional financing.
The foregoing sets forth many, but not all, of the factors that could cause actual results to differ from our expectations in any forward-looking statement. When evaluating our forward-looking statements, you should consider this cautionary statement along with the sections entitled “Risk Factor Summary,” “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Quantitative and Qualitative Disclosures about Market Risk” in this Annual Report on Form 10-K, as well as the risks identified in our other reports filed with the U.S. Securities and Exchange Commission (the "SEC"). New factors may emerge from time to time, and it is not possible for management to predict all such factors, nor can it assess the impact of any such factor on the business or the extent to which any factor, or combination of factors, may cause results to differ materially from those contained in any forward-looking statement.
NOTE REGARDING COMPANY REFERENCES
References in this report to “Emergent,” the “Company,” “we,” “us,” and “our” refer to Emergent BioSolutions Inc. and its consolidated subsidiaries.
NOTE REGARDING TRADE NAMES
Emergent®, BioThrax®, BaciThrax®, BAT®, ANTHRASIL®, CNJ-016®, ACAM2000®, NARCAN®, CYFENDUS®, TEMBEXA® and any and all Emergent BioSolutions Inc. brands, products, services and feature names, logos and slogans are trademarks or registered trademarks of Emergent BioSolutions Inc. or its subsidiaries in the United States or other countries. All other brands, products, services and feature names or trademarks are the property of their respective owners, including KLOXXADO®, which is a registered trademark of Hikma Pharmaceuticals USA Inc.
2
PART I
ITEM 1. BUSINESS
OVERVIEW
We are a global life sciences company focused on providing innovative preparedness and response solutions addressing accidental, deliberate and naturally occurring Public Health Threats (“PHTs”). Our solutions include a product portfolio, a product development portfolio, and a contract development and manufacturing (“CDMO”) services portfolio. The types of PHTs we are currently addressing are focused on the following four categories:
•chemical, biological, radiological, nuclear and explosives (“CBRNE”);
•emerging infectious diseases (“EID”);
•emerging health crises; and
•acute, emergency and community care.
Our revenues are derived from a combination of the sale and procurement of our product/product candidate portfolio (described below), the provision of our bioservices to external customers, and non-dilutive contract and grant funding for research and development (“R&D”) projects from various third-party sources.
As of December 31, 2025, the Company has a portfolio of 11 products focused on addressing global public health threats like smallpox, mpox, anthrax, Ebola and opioid overdose emergencies. The revenues generated by the products comprise a substantial portion of our revenue. We structure our business with a focus on markets and customers. As such, the key components of the business structure include the following four product and service categories: Commercial products, consisting of NARCAN® Nasal Spray and KLOXXADO®, Anthrax – Medical Countermeasures (“MCM”) Products, Smallpox - MCM products, and Emergent Bioservices (CDMO) (“Bioservices”).
OUR OPERATING SEGMENTS
Our business is organized in three operating segments:
•our Commercial Product Segment consisting of NARCAN® Nasal Spray and KLOXXADO® (naloxone HCl) Spray;
•our MCM Products Segment consisting of Anthrax—MCM products, Smallpox—MCM products and Other Products (as discussed below); and
•our Services Segment consisting of our Bioservices portfolio.
Additionally, we have a centralized R&D organization and an enterprise-wide governance approach to managing our portfolio of R&D projects.
Commercial Product Segment
In the U.S. and Canadian markets, our Commercial business primarily focuses on sales of naloxone products, NARCAN® (naloxone HCl) Nasal Spray and KLOXXADO® (nalonxone HCI) Nasal Spray. NARCAN® Nasal Spray is sold commercially over-the-counter ("OTC") at retail pharmacies and digital commerce websites as well as through physician-directed or standing order prescriptions at retail pharmacies, health departments, local law enforcement agencies, community-based organizations, substance abuse centers and other federal agencies. KLOXXADO® Nasal Spray is a prescription pharmaceutical product sold at pharmacies as well as public and government entities through physician-directed or standing order prescriptions.
3
In 2023, we completed the sale of our Commercial Products Segment's travel health business, including rights to Vivotif®, the licensed typhoid vaccine; Vaxchora®, the licensed cholera vaccine; the development-stage chikungunya vaccine candidate CHIKV VLP; the Company’s manufacturing site in Bern, Switzerland; and certain of its development facilities in San Diego, California to Bavarian Nordic. We have achieved all four development milestones under the Purchase and Sale Agreement with Bavarian Nordic for aggregate payments of $80.0 million: $10.0 million upon Bavarian Nordic's announcement that the European Medicines Agency had validated the marketing authorization application for CHIKV VLP in July 2024; $20.0 million upon Bavarian Nordic's announcement that the FDA accepted and granted Priority Review for the Biologics License Application ("BLA") for CHIKV VLP in August 2024; $30.0 million, upon the FDA's approval of CHIKV VLP under the Priority Review for persons 12 years of age and older in February 2025; and $20.0 million upon the European Commission's approval of CHIKV VLP for persons 12 years of age and older in February 2025. As a result, the entire $80.0 million of total milestone payments have been received as of December 31, 2025. Pursuant to the Purchase and Sale Agreement, the Company may receive up to $30.0 million of earn-out payments from Bavarian Nordic based on aggregate net sales of Vaxchora® and Vivotif® in calendar year 2026.
In connection with the divestiture, the Company entered into a Transition Services Agreement with Bavarian Nordic to help support its ongoing operations that was substantially completed in 2024. For additional information, refer to Note 4, "Divestitures" in the Notes to Consolidated Financial Statements in Part II, Item 8. of this Form 10-K.
MCM Products Segment
Our Government - MCM business focuses primarily on procurement of MCM products and procured product candidates by domestic and international government customers, with an emphasis on the USG, which is our largest customer. We also sell MCM products and procured product candidates to domestic and international non- and quasi-government organizations and to governments outside of the U.S.
In July 2024, we entered into a Stock and Asset Purchase Agreement (the “RSDL® Agreement”) with SERB Pharmaceuticals, through its wholly owned subsidiary BTG International Inc. (collectively, “SERB”), pursuant to which, among other things, we sold our worldwide rights to RSDL® to SERB (the “RSDL® Transaction”). See Note 4, “Divestitures” in the Notes to Consolidated Financial Statements in Part II, Item 8. of this Form 10-K for more information on the RSDL® Transaction.
Services Segment
Our services business consists of distinct but interrelated bioservices based on our manufacturing capabilities: drug substance manufacturing; drug product manufacturing (also referred to as “fill/finish” services) and packaging; development services including technology transfer, process and analytical development services; and, when necessary, suite reservation obligations. Our services utilize diverse technology platforms (mammalian, microbial, viral and plasma) from our development and manufacturing facilities to support internal products and enable us to offer unique capabilities to both clinical-stage and commercial-stage projects for a variety of third-party customers, including government agencies, innovative pharmaceutical companies, and non-government organizations.
Following the completion of multiple restructuring plans from January 2023 through September 2025, along with the sale of the Baltimore-Bayview drug substance manufacturing facility to Syngene International (“Syngene”), closure of the Rockville, Maryland Drug Product facility, and sale of Drug Product facility in Baltimore-Camden to Bora Pharmaceuticals Injectables Inc., a subsidiary of Bora, we successfully reduced investment in and de-emphasized focus on the Services business. Therefore, as of the first quarter of 2025, the Company’s Services operating segment no longer met the quantitative thresholds of a reportable segment and did not meet the aggregation criteria set forth in Accounting Standards Codification 280, Segment Reporting, and as such is categorized within “All other revenues” along with “Contracts and Grants”. See Note 19 “Segment information”, Note 4, “Divestitures” and Note 5, “Impairment and restructuring charges” in the Notes to Consolidated Financial Statements in Part II, Item 8. of this Form 10-K for more information related to these announcements.
OUR STRATEGY
In 2025, the Company advanced the turnaround phase of its multi-year plan while remaining focused on our mission to protect and save lives. We delivered solutions for high-priority public health threats and opioid overdose emergencies and executed key actions to improve operating performance, strengthen financial flexibility, streamline our manufacturing footprint, maintain leadership in naloxone, and deliver solid performance in our MCM business globally.
4
Early in the year, we took meaningful steps to position the Company for long-term organic and inorganic growth and to further strengthen our financial profile. In March 2025, we completed the sale of the Baltimore-Bayview drug substance manufacturing facility to Syngene for approximately $36.5 million at closing, while retaining the right to secure manufacturing services and capacity at the facility for future growth and pandemic response production in collaboration with Syngene. The Company also expanded its naloxone portfolio through the acquisition from Hikma of exclusive commercial rights in the U.S. and Canada to KLOXXADO® (naloxone HCl) Nasal Spray 8 mg. We also progressed a strategic innovation initiative through investment in Swiss Rockets Ltd. During the year, the Company also undertook several actions to support value creation for shareholders. In March 2025, our Board of Directors authorized a stock repurchase program of up to $50.0 million through March 27, 2026, and authorized the Company to repurchase up to $30.0 million in aggregate principal amount of the 3.875% Senior unsecured Notes due 2028. Taken together with the Company's voluntary $100.0 million prepayment under the Company's Term Loan Agreement with OHA Agency LLC, as administrative agent, and the lenders from time to time party thereto (the “Term Loan Agreement”) in late December 2025. In February 2026, our Board of Directors reauthorized the 2025 Share Repurchase Program for the repurchase of up to $50.0 million through March 31, 2027. These actions demonstrate our continued focus on a disciplined capital allocation strategy and enhanced financial flexibility.
Operationally the Company continued to streamline and strengthen our manufacturing network and execution capabilities, driving improved utilization and profit follow-through. In December 2025, the FDA approved a supplemental BLA to add our Winnipeg, Canada facility as the drug product manufacturing and testing site for raxibacumab, supporting our objective of a more resilient, customer-focused network capable of supplying critical products and supporting strategic manufacturing partnerships.
The Company maintained strong collaboration with U.S. government partners and continued to meet expectations for MCM preparedness, securing multiple contract actions during 2025 that support near- and mid-term deliveries. These included contract actions for ACAM2000® totaling $56 million, CYFENDUS® totaling $30 million, CNJ-016® (VIGIV) totaling $51.9 million, BAT® totaling $62.4 million, and TEMBEXA® totaling $17 million. The Company also announced the exercise of a $20 million option to supply BioThrax® (Anthrax Vaccine Adsorbed) to the U.S. Department of War and received a Biomedical Advanced Research and Development Authority ("BARDA") contract option valued at approximately $16.7 million to continue post-licensure development collaboration activities for Ebanga™.
Internationally, the Company continued to pursue opportunities aligned with our capabilities and partners’ preparedness needs. During 2025, we announced approximately $27 million in incremental sales to supply international customers with five critical MCM products, and we secured $29 million in MCM product orders from an international government partner.
As biological threats continue to evolve, we have also engaged stakeholders to reinforce the importance of sustained preparedness and public-private partnership, including communications intended to elevate awareness of biological threats and U.S. and global preparedness needs.
The Company continued to hold a leadership position with NARCAN® Nasal Spray, supported by strong demand for OTC NARCAN® Nasal Spray and continued innovation in customer solutions. We expanded NARCANDirect® to offer KLOXXADO® and introduced Convenience Kits to support frontline responders and community programs, while continuing to invest in overdose preparedness and awareness initiatives intended to normalize carrying naloxone and reduce stigma.
Looking to 2026, the Company plans to continue executing the turnaround phase of our multi-year plan with a focus on disciplined execution, improved operating performance, and long-term sustainable growth while maintaining our mission to protect and save lives. The priorities include:
•Reinforcing the highest standards of patient safety, quality and compliance,
•Leveraging strong government and stakeholder relationships to support preparedness priorities,
•Maintaining disciplined capital allocation and financial flexibility,
•Enabling growth across our business lines and strengthening our competitive positioning,
•Advancing strategic partnerships and targeted innovation aligned to our capabilities, and
•Expanding global engagement to support international opportunities.
5
PRIMARY PRODUCTS AND PRODUCT CANDIDATES
Government - MCM Products
The current portfolio of our Government - MCM business consists of the following products:
GOVERNMENT - MCM PRODUCTS | ||||||||
Product | Indication(s)* | Regulatory Approvals, Licensures or Clearances | ||||||
ACAM2000® (Smallpox (Vaccinia) Vaccine, Live) | Vaccine for active immunization against smallpox disease for persons determined to be at high risk for smallpox infection.** | Australia, Canada, Singapore | ||||||
ACAM2000® (Smallpox and Mpox (Vaccinia) Vaccine, Live) | Vaccine for active immunization for the prevention of smallpox and mpox disease in individuals determined to be at high risk for smallpox or mpox infection. | United States | ||||||
ANTHRASIL® [Anthrax Immune Globulin Intravenous (Human)] | Treatment of inhalational anthrax in adult and pediatric patients in combination with appropriate antibacterial drugs. | United States, Canada | ||||||
BAT® [Botulism Antitoxin Heptavalent (A,B,C,D,E,F,G)-(Equine)] | Treatment of symptomatic botulism following documented or suspected exposure to botulinum neurotoxin serotypes A, B, C, D, E, F, or G in adults and pediatric patients. | United States, Canada, Singapore, Ukraine | ||||||
BioThrax® (Anthrax Vaccine Adsorbed) | Vaccine for active immunization for the prevention of disease caused by Bacillus anthracis in persons 18 through 65 years of age. BioThrax® is approved for: 1.Pre-exposure prophylaxis of disease in persons at high risk of exposure. 2.Post-exposure prophylaxis of disease following suspected or confirmed Bacillus anthracis exposure, when administered in conjunction with recommended antibacterial drugs. | United States, Canada, France (where it is known as BaciThrax®), Germany, Singapore | ||||||
CYFENDUS® (Anthrax Vaccine Adsorbed Adjuvanted) | Vaccine for post-exposure prophylaxis of disease following suspected or confirmed exposure to Bacillus anthracis in persons 18 through 65 years of age when administered in conjunction with recommended antibacterial drugs. | United States | ||||||
Ebanga™ (ansuvimab-zykl), for injection | Treatment of infection caused by Orthoebolavirus zairense in adult and pediatric patients, including neonates born to a mother who is RT-PCR positive for Orthoebolavirus zairense infection. | United States | ||||||
| Raxibacumab injection | Treatment of adult and pediatric patients with inhalational anthrax due to Bacillus anthracis in combination with appropriate antibacterial drugs and for prophylaxis of inhalational anthrax when alternative therapies are not available or are not appropriate. | United States | ||||||
TEMBEXA® (brincidofovir), (tablet and oral suspension) | Treatment of human smallpox disease caused by variola virus in adult and pediatric patients, including neonates. | United States, Canada | ||||||
CNJ-016® (VIGIV) [Vaccinia Immune Globulin Intravenous (Human)] | Treatment of complications due to vaccinia vaccination, including: •Eczema vaccinatum •Progressive vaccinia; •Severe generalized vaccinia; •Vaccinia infections in individuals who have skin conditions; and •Aberrant infections induced by vaccinia virus (except in cases of isolated keratitis). CNJ-016® (VIGIV) is not indicated for postvaccinial encephalitis. | United States, Canada | ||||||
*Indication statements are as per the U.S. Prescribing Information unless otherwise denoted. **Indication statement as per the Health Canada Product Monograph. | ||||||||
6
Description of MCM Products
ACAM2000® (Smallpox and Mpox (Vaccinia) Vaccine, Live). ACAM2000® vaccine is a smallpox and mpox vaccine licensed by the FDA for active immunization for the prevention of smallpox and mpox disease in individuals determined to be at high risk for smallpox or mpox infection. The mpox indication was approved by the FDA following a supplemental Biological License Application ("sBLA") approval issued in August 2024. In December 2023, ACAM2000® vaccine received a Notice of Compliance from Health Canada for its Extraordinary Use New Drug Submission (EUNDS) for the indication of active immunization against smallpox disease for persons determined to be at high risk for smallpox infection. ACAM2000® is also licensed in Australia and Singapore for the smallpox indication. ACAM2000® vaccine is currently stockpiled both in the U.S. and internationally. Smallpox is a highly contagious disease caused by the Variola virus. According to the Centers for Disease Control and Prevention ("CDC"), smallpox is a devastating disease, with a mortality rate as high as 30%. In recent years, there have been global outbreaks of mpox, which is caused by a virus in the same family as smallpox, increasing the need for vaccine access. This is particularly true in endemic regions. The vaccine stimulates a person's immune system to develop antibodies and cell responses that can then help the body fight off infection if exposure occurs.
In 2019, we announced the award by the USG of a contract valued at up to approximately $2.0 billion over 10 years for the continued supply of ACAM2000® vaccine into the SNS, assuming all contract options are exercised (the "ACAM2000® Contract"). This multiple-year contract is intended to support the replacement of the smallpox vaccine stockpile and included a one-year base period of performance in 2019 valued at approximately $170.0 million, and nine option years. The number of doses under the base period were delivered by year-end 2019. The U.S. Department of Health and Human Services ("HHS") exercises its first and second contract options to procure doses of ACAM2000® vaccine in 2020 and 2021, respectively, which options were valued at $176.0 million and $182.2 million, respectively. The USG chose to not exercise its option year in 2022. In 2023, 2024 and 2025, the Company, through its wholly-owned subsidiary, Emergent Product Development Gaithersburg Inc. ("Emergent Gaithersburg") received contract modifications from the Office of the Assistant Secretary for Preparedness and Response (“ASPR”), an agency of HHS, exercising and funding the third, fifth and sixth, respectively, of nine annual contract term extension options (the “Option Exercises”) for Emergent to supply ACAM2000® to the SNS. These option exercises were valued at approximately $120.0 million, $99.9 million and $56.0 million, respectively. The Option Exercises were made under bilateral modifications of the ACAM2000® Contract. All delivery options were satisfied when met.
ANTHRASIL®. ANTHRASIL® (Anthrax Immune Globulin Intravenous (Human)) is the only polyclonal antibody therapeutic licensed by the FDA for the treatment of inhalational anthrax in adult and pediatric patients in combination with appropriate antibacterial drugs. In 2024, we continued two contracts with HHS for ANTHRASIL®: 1) a development and procurement contract which was successfully completed as of September 2024, and 2) an active delivery order issued under a multiple award, indefinite delivery/indefinite quantity contract. The delivery order was successfully completed in July 2025 with formal close out executed in September 2025. In addition to domestic USG revenue, ANTHRASIL® has been sold to several foreign governments.
BAT® [Botulism Antitoxin Heptavalent (A, B, C, D, E, F, G) – (Equine)]. BAT® antitoxin is the only equine plasma antitoxin licensed by the FDA and Health Canada for the treatment of all seven botulinum neurotoxin serotypes. BAT® antitoxin is licensed by the FDA for the treatment of symptomatic botulism following suspected or documented exposure to botulinum neurotoxin serotypes A, B, C, D, E, F or G in adults and pediatric patients. It is also licensed in Canada pursuant to Health Canada’s Extraordinary Use New Drugs regulations. BAT® antitoxin is the only heptavalent botulism antitoxin available in the U.S. and Canada for treating naturally occurring botulism in adults or pediatric patients. Botulinum toxin is a nerve toxin produced by the bacterium Clostridium botulinum that causes botulism, a serious paralytic illness. In 2020, we announced the finalization of a previously announced contract with HHS, valued at up to $550.0 million, if all options under the contract are exercised. The contract has two deliverables. The first deliverable, negotiated in September 2019 and valued at up to approximately $90.0 million, is to supply annual doses of BAT® antitoxin into the SNS for 10 years by converting existing bulk drug substance into final drug product. This deliverable also includes options for additional doses valued at up to approximately $94.0 million over 10 years. The second deliverable, valued at up to approximately $366.0 million, is for the production of additional doses of bulk drug substance over 10 years to maintain the plasma collection and production capability for botulism response planning. In addition to domestic government sales, BAT® antitoxin continues to be sold internationally, with deliveries to 19 foreign countries in 2025.
7
BioThrax® (Anthrax Vaccine Adsorbed). BioThrax® vaccine is the only vaccine licensed by the FDA for pre-exposure prophylaxis of anthrax disease in persons at high risk of exposure. BioThrax® vaccine is also approved by the FDA for post-exposure prophylaxis of disease following suspected or confirmed Bacillus anthracis exposure, when administered in conjunction with recommended antibacterial drugs. Anthrax is a potentially fatal disease caused by the spore-forming bacterium, Bacillus anthracis. Inhalational anthrax is the most lethal form of anthrax. In the U.S., BioThrax® vaccine is administered in a pre-exposure prophylaxis setting by intramuscular injection as a three-dose primary series over a six-month period. Per the U.S. label, booster doses are administered six and 12 months after completion of the primary series and at 12-month intervals thereafter. BioThrax® vaccine is administered in a post-exposure prophylaxis setting as three subcutaneous injections two weeks apart in conjunction with antibacterial drugs following suspected or confirmed Bacillus anthracis exposure. When we report the revenue associated with “anthrax vaccines,” it reflects the combined revenue from the procurement and sale of BioThrax® vaccine as well as CYFENDUS® vaccine.
In January 2024, the Company announced that it has secured an indefinite-delivery, indefinite-quantity ("IDIQ") procurement contract with the U.S. Department of War ("DoW") and led by the Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense, with a maximum value up to $235.8 million, to supply BioThrax® vaccine for use by all branches of the U.S. military as Pre-Exposure Prophylaxis (PrEP) for anthrax disease. The IDIQ contract is comprised of a five-year base agreement ending on September 30, 2028, and an additional five-year option that would extend the contract to September 30, 2033. In 2024, one delivery order was issued, exercising two contract options for $27.9 million. In January 2025, the Company announced the exercise of a third option and modification to the IDIQ contract valued at approximately $20 million to supply BioThrax® vaccine for use by all branches of the U.S. military as Pre-Exposure Prophylaxis (PrEP) for anthrax disease. On January 8, 2026, the Company announced the exercise of an option to the IDIQ contract valued at approximately $21.5 million to supply BioThrax® to the U.S. Department of War in 2026.
CYFENDUS® (Anthrax Vaccine Adsorbed, Adjuvanted) (referred to as investigational product AV7909 prior to FDA approval in July 2023). CYFENDUS® vaccine was approved by the FDA for post-exposure prophylaxis of disease following suspected or confirmed exposure to Bacillus anthracis in persons 18 through 65 years of age when administered in conjunction with recommended antibacterial drugs. In 2021, AV7909 was granted orphan drug designation by the FDA. In 2023, the FDA approved our BLA for CYFENDUS®. Studies have shown that CYFENDUS® elicits a stronger immune response using fewer doses than BioThrax® vaccine, which is expected to allow patients to reach a protective level of immunity more rapidly. CYFENDUS® is designed to provide protection with a two-dose regimen (versus the BioThrax® three-dose regimen) for post-exposure prophylaxis of anthrax disease, when administered in combination with the recommended antibacterial drugs.
In 2016, we signed a combination development and procurement contract with Biomedical Advanced Research and Development Authority ("BARDA"), which included a five-year base period of performance to develop CYFENDUS® for post-exposure prophylaxis of anthrax disease and to deliver to the SNS an initial two million doses, subsequently modified to three million doses in March 2017. The contract also includes procurement options for the delivery of an additional 7.5 million to 50.0 million doses of CYFENDUS® into the SNS and options for an additional clinical study and post marketing commitments. In 2019, we initiated and completed enrollment of a Phase 3 study of 3,850 subjects evaluating safety, immunogenicity and lot consistency. The study was completed in 2020. In collaboration with us, the CDC filed with the FDA a pre-EUA submission package related to CYFENDUS®. Following this submission, BARDA began procuring CYFENDUS®, exercising its first three contract options in July 2019, July 2020 and September 2021, respectively, followed by the execution of two contract modifications in 2023. In 2024, two additional contract modifications (valued at approximately $80.0 million) were executed with BARDA; full delivery under the first modification was completed in 2024, and partial delivery for the second modification occurred in 2024 with the remainder to be delivered in 2025.
EbangaTM (ansuvimab-zykl), for injection. EbangaTM (ansuvimab-zykl) is a monoclonal antibody with antiviral activity provided through a single IV infusion (over 60 minutes) for the treatment of infection caused by Orthoebolavirus zairense in adult and pediatric patients, including neonates born to a mother who is RT-PCR positive for Orthoebolavirus zairense infection. In 2022, we entered into an asset purchase agreement and a license agreement with Ridgeback Biotherapeutics (“Ridgeback”) to expand the availability of EbangaTM (ansuvimab-zykl). This included a license to use the Ebanga™ trademark and to certain patent rights related to EbangaTM (ansuvimab-zykl). We are responsible for manufacturing, selling and distributing EbangaTM (ansuvimab-zykl) in the U.S. and Canada and Ridgeback will serve as the global access partner. In 2023, we announced we were awarded a 10-year contract by BARDA, valued at up to a maximum of $704 million, for advanced development, manufacturing scale-up, and procurement of Ebanga™. In 2024, BARDA elected to exercise Option 1 (valued at approximately $41.9 million) under the contract for work related to drug substance engineering, scale-up, stability, process validation and commercial readiness. In January 2025, BARDA elected to exercise Option 2 (valued at approximately $16.7 million) under the contract for work related to drug product process and analytical testing validation and long-term stability for Ebanga™ (ansuvimab-zykl).
8
Raxibacumab injection. Our raxibacumab product is the first fully human monoclonal antibody therapeutic licensed by the FDA for the treatment and prophylaxis of inhalational anthrax due to Bacillus anthracis. Our raxibacumab product is indicated for the treatment of adult and pediatric patients with inhalational anthrax due to Bacillus anthracis in combination with appropriate antibacterial drugs and for prophylaxis of inhalational anthrax when alternative therapies are not available or are not appropriate.
TEMBEXA® (brincidofovir) tablets and oral suspension. TEMBEXA® is the first oral antiviral approved by the FDA for the treatment of smallpox disease caused by variola virus in adult and pediatric patients, including neonates. In September 2022, we acquired exclusive worldwide rights to brincidofovir from Chimerix Inc. for the treatment of any human smallpox disease or any other disease caused by any orthopox virus. Following the acquisition, the 10-year contract with BARDA, valued at up to $680.0 million, to supply up to 1.7 million tablet and suspension formulations of TEMBEXA® was novated to the Company, which contract was modified in 2024 to execute additional procurement options valued at approximately $67.4 million. In September 2025, an option was exercised for additional procurement of TEMBEXA® valued approximately at $17.1 million. In 2023, Emergent received a Notice of Compliance from Health Canada for its Extraordinary Use New Drug Submission (EUNDS) for indication of the treatment of human smallpox disease in adults and pediatric patients, including newborn infants. In 2024, Emergent announced that TEMBEXA® would be included in a clinical trial conducted and sponsored by PANTHER, under the leadership of the Africa Centers for Disease Control and Prevention, as part of the Mpox Study in Africa (MOSA). The Company has two patent families relating to the TEMBEXA® product including but not limited to, those listed in the Orange Book: 8,962,829, 9,303,051, 9,371,344, 10,112,909, and 10,487,061. The latest expiring United States composition of matter patents expires in 2034.
CNJ-016® (VIGIV) (Vaccinia Immune Globulin Intravenous (Human)). CNJ-016® (VIGIV) is the only polyclonal antibody therapeutic licensed by the FDA and Health Canada to address certain complications from replicating virus smallpox vaccination. The principal customer for CNJ-016® (VIGIV) is the USG, specifically HHS. In 2019, we announced a contract award by HHS valued at approximately $535.0 million over 10 years for the continued supply of CNJ-016® (VIGIV) into the SNS for smallpox preparedness. CNJ-016® (VIGIV) has also been procured by a limited number of foreign governments.
Commercial Products
Our current commercial portfolio consists of the following product:
COMMERCIAL PRODUCTS | ||||||||
Product | Indication(s)* | Regulatory Approvals | ||||||
NARCAN® (naloxone HCI) Nasal Spray | Emergency use to reverse known or suspected opioid overdose as manifested by respiratory and/or severe central nervous system depression. | United States, Canada | ||||||
KLOXXADO® (naloxone HCI) Nasal Spray | Emergency treatment of known or suspected opioid overdose as manifested by respiratory and/or central nervous system depression, for adult and pediatric patients.** | United States, Canada | ||||||
*Indication statement as per the Health Canada Product Monograph **Indication statement is as per the U.S. Prescribing Information | ||||||||
Description of Commercial Products
NARCAN® Nasal Spray. NARCAN® Nasal Spray, a product we obtained in connection with our acquisition of Adapt Pharma Inc. in 2018, is an intranasal formulation of naloxone approved by the FDA and Health Canada for the emergency treatment of known or suspected opioid overdose as demonstrated by respiratory and/or central nervous system depression. We have rights to formulations of naloxone used in NARCAN® Nasal Spray from Indivior PLC (f/k/a Opiant Pharmaceuticals Inc.). The primary customers for NARCAN® Nasal Spray are state health departments, local law enforcement agencies, community-based organizations, substance abuse centers, federal agencies, and consumers. We launched the Generation II NARCAN® Nasal Spray device, which has a claim for enhanced temperature excursions and storage below 25°C. In January 2024, the Company announced that the FDA has acknowledged the shelf-life extension of NARCAN® Nasal Spray from 36 months to 48 months based on the Company’s four-year stability data.
In February 2023, the FDA Nonprescription Drugs Advisory Committee and the Anesthetic and Analgesic Drug Products Advisory Committee unanimously voted in favor (a total of 19 votes) that the benefit-risk profile of NARCAN® Nasal Spray was supportive of its use as a nonprescription opioid overdose reversal agent. NARCAN® Nasal Spray was approved as an OTC medication in March 2023. It was the first 4 mg naloxone nasal spray available OTC in the U.S. The OTC product was shipped out to retailers and e-commerce providers nationwide beginning in August 2023 and continues to be available nationwide across grocery, drug stores and mass merchant.
9
In January 2025, the Company announced an agreement with Hikma Pharmaceuticals Inc. (“Hikma”) in which the Company obtained exclusive commercial rights for product sales and marketing in the United States and Canada to Hikma’s KLOXXADO® (Naloxone HCl) Nasal Spray, for the emergency treatment of known or suspected opioid overdose as demonstrated by respiratory and/or central nervous system depression. KLOXXADO® is a prescription product initially approved with 8mg dose in the U.S. and Canada, and a 4mg dose additionally approved in the United States in 2025. Emergent took over sales and marketing of KLOXXADO® in 2025. The primary customers for KLOXXADO® are state health departments, local law enforcement agencies, community-based organizations, substance abuse centers, federal agencies, and patients with prescriptions accessing the product via pharmacies.
Product Candidates
The table below highlights our product candidates in 2025:
PRODUCT CANDIDATES | ||||||||
Product Candidate | Target Indication | Stage of Development | ||||||
| EBS-MARV (rVSV-vectored vaccine for Marburg virus disease) | Active immunization to prevent Marburg virus disease. | Pre-Clinical | ||||||
| EBS-SUDV (rVSV-vectored vaccine for Sudan virus disease) | Active immunization to prevent Sudan virus disease. | Pre-Clinical | ||||||
| Pan-Ebola mAbs | Treatment of Ebola virus disease caused by infection in patients with confirmed Sudan virus. | Pre-Clinical | ||||||
| WEVEE-VLP | To prevent disease caused by Western, Eastern, and Venezuelan equine encephalitis virus infections. | Phase 1 - Completed | ||||||
Description of Product Candidates
EBS-LASV. This vaccine candidate is a recombinant, vesicular stomatitis virus vectored, monovalent vaccine encoding the surface glycoprotein precursor gene of Lassa virus. The development program is partnered with the Coalition for Epidemic Preparedness Innovations ("CEPI"). We have completed our Phase 1 clinical trial and while no safety concerns were observed, the vaccine did not meet the specified immunogenicity endpoint. CEPI funding has been discontinued and this candidate will not progress into Phase 2. This program is discontinued and was closed in 2025.
EBS-MARV. This vaccine candidate is a recombinant, vesicular stomatitis virus vectored, monovalent vaccine encoding the surface glycoprotein precursor gene of Marburg virus. The development program is partnered with Auro Vaccines and is currently in IND-enabling stage. National Institute of Allergy and Infectious Diseases ("NIAID") is funding Auro Vaccines program with contract options through Phase 1 development. Phase 1 GMP Drug Substance material was manufactured in 2025. Discussions are on-going between Auro Vaccines and NIAID around funding for conversion of this material to GMP Drug Product. At the end of 2025, the Company decided to deprioritize this product candidate.
EBS-SUDV. This vaccine candidate is a recombinant, vesicular stomatitis virus vectored, monovalent vaccine encoding the surface glycoprotein precursor gene of Sudan virus. The development program is partnered with Auro Vaccines and is currently in IND-enabling stage. NIAID is funding Auro Vaccines program with contract options through Phase 1 development. Phase 1 GMP material was not manufactured as expected in 2025 due to NIAID funding challenges. At the end of 2025, the Company decided to deprioritize this product candidate.
Pan-Ebola mAbs. IBT-T02 is a cocktail comprised of two chimeric mAbs, FVM04 and CA45. The mAb cocktail candidate targets two highly conserved and non-overlapping epitopes on the glycoprotein of Sudan ebolavirus (SUDV) and other ebolaviruses. The preclinical program is in partnership with Integrated Biotherapeutics. The BARDA funded non-human primate study was completed and the data were reported in 2025. At the end of 2025, the Company decided to deprioritize this product candidate.
WEVEE-VLP. WEVEE VLP vaccine is a recombinant VLP vaccine. The development program is in partnership with NIAID Vaccine Research Center (VRC), who has already completed a Phase 1 clinical trial of the trivalent vaccine, in addition to a monovalent Phase 1 clinical study of the VEEV VLP component only. At the end of 2025, the Company decided to deprioritize this product candidate.
10
Description of Services
Bioservices
Our Bioservices offering is based on our established development and manufacturing infrastructure, technology platforms and expertise related to the production of biopharmaceutical products.
Our Bioservices consist of development and analytical services, bulk drug substance manufacturing, drug product formulation and filling, and packaging of final drug product. Collectively, this portfolio of services provides the capability to support “molecule-to-market” solutions to clients engaged in drug development through commercialization of specific product types. These services are provided to biopharmaceutical innovator companies and non-governmental organizations ("NGOs").
We currently have four development and manufacturing sites located in the U.S. and Canada. These sites allow us to meet our internal manufacturing needs as well as perform targeted services for our external customers.
•Our Winnipeg and Gaithersburg sites house our development services expertise;
•Our Canton, Lansing and Winnipeg sites house our commercial drug substance expertise; and
•Our Winnipeg site houses our drug product and packaging expertise.
Marketing and Sales
We have dedicated sales channels for each of our products and service offerings.
Government - MCM Products
Our dedicated team possesses specialized knowledge and experience in both the public and private sectors, focusing on counter terrorism, CBRNE preparedness and public health. We facilitate the entire procurement process for our MCM products and procured product candidates globally through partnerships with the USG, international and domestic NGOs and foreign governments.
The following table lists the registered trademarks for our MCM products:
Trademark | Country of Origin | ||||
ACAM2000® | Australia, Brazil, Canada, EU, Hong Kong, Israel, Singapore, United Kingdom, U.S. | ||||
ANTHRASIL® | Australia, Canada, Egypt, EU, Israel, Lebanon, Republic of Korea, Qatar, Saudi Arabia, Singapore, Turkey, United Kingdom, United Arab Emirates, U.S. | ||||
 | U.S. | ||||
 | EU, UK | ||||
BAT® | Canada, EU, Japan, Mexico, United Kingdom, U.S. | ||||
BioThrax® | Australia, Brazil, Canada, EU, India, Malaysia, Saudi Arabia, Singapore, United Kingdom, U.S. | ||||
BaciThrax® | France | ||||
CYFENDUS® | U.S. | ||||
TEMBEXA® | Argentina, Australia, Brazil, Canada, Chile, China, EU, Hong Kong, India, Israel, Japan, Mexico, New Zealand, Norway, Russia, Singapore, South Africa, Switzerland, Taiwan, Turkey, Ukraine, United Kingdom, U.S. | ||||
 | Australia, EU | ||||
CNJ-016® | U.S. | ||||
11
Commercial Products
In the U.S. market, NARCAN® (naloxone HCl) Nasal Spray and KLOXXADO® are sold directly to state and local governments and used by first responders, including: police, firefighters and emergency medical teams. In addition, NARCAN® Nasal Spray is sold to consumers online and through retailers nationwide, as well as to businesses. NARCAN® Nasal Spray was approved as an OTC medication in March 2023. It was the first 4 mg naloxone nasal spray available OTC in the U.S. KLOXXADO®, a prescription product, is also available to patients with prescriptions at pharmacies.
In December 2024, Emergent completed its FDA establishment registration (per Title 21 CFR Part 807) to authorize Emergent to distribute imported convenience kits containing medical devices (gloves and CPR mask) in the U.S. These convenience kits provide devices that support treatment of opioid overdose.
In the Canadian market, NARCAN® (naloxone HCl) Nasal Spray is sold directly to federal, provincial and local governments and agencies for use by first responders, public health and harm reduction agencies, and indigenous communities. In addition, NARCAN® Nasal Spray is sold to businesses online and through pharmacies nationwide. In November 2023, we submitted a Medical Device Establishment License (MDEL) Application to Health Canada that would permit Emergent to distribute naloxone convenience kits containing medical devices (gloves and CPR mask) and Narcan® Nasal Spray (4mg) to comply with provincial legislative requirements. In December 2023, Emergent Canada Inc. received the MDEL, enabling the direct distribution of naloxone convenience kits containing medical devices and NARCAN® Nasal Spray in Canada, which distribution was launched in 2024.
12
The following table lists the registered trademarks for NARCAN® (naloxone HCl) Nasal Spray:
Trademark | Country of Registration | ||||
NARCAN® | Benelux, Canada, Denmark, Estonia, EU, Finland, Germany, Ireland, Italy, Norway, Spain, Sweden, United Kingdom, U.S. | ||||
 | Canada, EU, Norway, United Kingdom, U.S. | ||||
| NarcanNasalSpray | Benelux, Germany, Denmark, Spain, Estonia, Finland, United Kingdom, Ireland, Italy, Norway, Sweden | ||||
| NARCAN TWINDOSE | Canada, EU, Norway, United Kingdom | ||||
| NARCAN DIRECT | Canada, EU, United Kingdom, U.S. | ||||
| LAY, SPRAY, STAY | EU, United Kingdom, U.S. | ||||
 | Canada, U.S. | ||||
 | Canada, U.S., EU, United Kingdom | ||||
 | Canada | ||||
 | Canada | ||||
 | Canada | ||||
 | EU, United Kingdom, U.S. | ||||
 | EU, United Kingdom | ||||
 | EU, United Kingdom, U.S. | ||||
 | U.S. | ||||
 | U.S. | ||||
13
Bioservices
We market our bioservices to the global pharmaceutical and biotechnology industry, governments and NGOs. Our bioservices are supported by a dedicated group of professionals qualified to represent the full breadth of our service offerings.
Competition
Our products and any product or product candidate that we acquire or successfully develop and commercialize are likely to compete with current products and product candidates that are in development for the same indications. The competition for our products and product candidates includes the following:
•ACAM2000®. ACAM2000® vaccine, which is licensed by the FDA for smallpox and mpox, and is licensed in Australia, Canada and Singapore for smallpox, remains the primary smallpox vaccine stockpiled by the USG and offers key features for public health mass vaccination programs that are critical, including a single dose vaccination schedule and multi-dose vial presentation. ACAM2000® vaccine faces competition from JYNNEOSTM vaccine, which is licensed by the FDA for the prevention of smallpox and mpox disease in adults 18 years of age and older determined to be at high risk for smallpox or mpox infection. JYNNEOS® vaccine is also approved in Switzerland, Singapore and Mexico. JYNNEOS® vaccine is approved in Canada and in the EU and United Kingdom under the trade names IMVAMUNE® and IMVANEX®, respectively. ACAM2000® vaccine also faces competition from KM Biologics LC16m8 vaccine, which is approved in Japan for smallpox and mpox.
•CYFENDUS® and BioThrax®. CYFENDUS® and BioThrax® vaccines are currently procured, primarily by the USG, for prevention of anthrax disease. BioThrax® and CYFENDUS® vaccines are currently the only two anthrax vaccines approved by the FDA for prevention of anthrax disease, and CYFENDUS® and BioThrax® are the only anthrax vaccines procured by the USG for the SNS to date. We face potential future competition for the supply of anthrax vaccines if the USG chooses to procure alternative products or product candidates. GC Biopharma, Blue Willow Biologics/Porton Biopharma, and Greffex are each currently developing anthrax vaccine product candidates, which are in various stages of clinical development. Of these product candidates, GC Biopharma and Blue Willow Biologics have completed Phase 1 trials.
•BAT®. Our botulinum antitoxin immune globulin product is the only heptavalent antitoxin licensed by the FDA and Health Canada for the treatment of symptomatic botulism for all seven botulinum neurotoxin serotypes. Direct competition is currently limited.
•CNJ-016®. Our VIGIV product is the only therapeutic licensed by the FDA and Health Canada to address adverse events from smallpox vaccination with replicating virus smallpox vaccines. While direct competition in terms of the treatment of smallpox vaccination side effects is limited, SIGA has obtained EU approval for TPOXX® (tecovirimat), an oral therapy, for the treatment of complications following vaccination against smallpox. TPOXX® is currently procured by the USG for the SNS.
•EbangaTM (ansuvimab-zykl). A monoclonal antibody therapeutic approved by the FDA in December 2020 for the treatment of infection caused by Zaire Ebolavirus in adult and pediatric patients, including neonates born to RT-PCR+ mother for Zaire Ebolavirus infection. Ebanga faces competition from another monoclonal antibody, Inmazeb® (atoltivimab, maftivimab and odesivimab-ebgn), which was approved by the FDA in October 2020 with the same indication. Inmazeb® is currently procured by the USG for the SNS.
14
•Naloxone Commercial Products. NARCAN® (naloxone HCl) Nasal Spray is the first FDA-approved intranasal naloxone spray for the emergency reversal of opioid overdoses. KLOXXADO® (naloxone HCI) nasal spray is a prescription product for the same use. These products face generic competition. Teva Pharmaceuticals Industries Ltd. and its Canadian affiliate (collectively, "Teva") have generic versions of an intranasal naloxone spray based on NARCAN® Nasal Spray approved by the FDA and Health Canada. Teva launched its generic naloxone nasal spray in the U.S. in 2021. Padagis Pharmaceuticals also has a generic version of an intranasal naloxone spray based on NARCAN® Nasal Spray approved by the FDA and launched in the U.S. in 2023. In addition, in April 2024, the FDA approved an additional ANDA for nasal naloxone 4mg from Amneal Pharmaceuticals, Inc. Our naloxone products also face branded competition: RextovyTM, (naloxone HCL nasal spray 4 mg), a branded product developed by Amphastar Pharmaceuticals, Inc. which also has a naloxone injection product, ZimhiTM (naloxone), a branded injectable product developed by Adamis, and RiVive™ a 3mg naloxone nasal spray formulation intended for use in opioid overdose reversal by Harm Reduction Therapeutics. In May 2023, FDA granted approval of OPVEE® (nalmefene) Nasal Spray to Opiant Pharmaceuticals Inc, (now a wholly owned subsidiary of Indivior PLC), which was released in the market during fourth quarter of 2023. In April 2024, the FDA approved Rezenopy® (naloxone HCL nasal spray 10mg), a branded product manufactured by Summit Biosciences Inc. Our naloxone products may face additional generic and branded competition in the future.
•Raxibacumab and ANTHRASIL® [Anthrax Immune Globulin Intravenous (Human)]. Our raxibacumab product is the first FDA-licensed fully human anthrax monoclonal antibody therapeutic and ANTHRASIL® [Anthrax Immune Globulin Intravenous (Human)] is the only polyclonal antibody therapeutic licensed by the FDA and Health Canada for the treatment of inhalational anthrax in adult and pediatric patients in combination with appropriate antibacterial drugs. Elusys Therapeutics, Inc. has obtained FDA licensure for Anthim® (obiltoxaximab) injection, a monoclonal antibody indicated for the treatment and prophylaxis of inhalational anthrax. Obiltoxaximab is also approved in Canada.
•TEMBEXA® (brincidofovir). TEMBEXA® is the first oral antiviral approved by the FDA, in June 2021, for all age groups for the treatment of smallpox. In 2023, TEMBEXA® received approval by Health Canada. TEMBEXA® faces competition from TPOXX® (tecovirimat), an oral therapy for the treatment of smallpox disease that was approved by the FDA in July 2018 and is currently procured by the USG for the SNS. TPOXX® is also approved in Canada and the EU. In the EU and Japan, TPOXX® is indicated for the treatment of smallpox, mpox and cowpox, as well as the treatment of complications following vaccination against smallpox.
Bioservices
We also compete for bioservices with several biopharmaceutical product R&D organizations, contract manufacturers of biopharmaceutical products (CDMOs), other bioservices organizations, and university research laboratories.
Companies with which we compete to provide bioservices fall into three broad categories. First, contract manufacturers such as Thermo Fisher Scientific, Lonza, and FUJIFILM Biotechnologies, all of which offer end-to-end contract manufacturing services encompassing development, drug substance, and drug product. Second, contract manufactures such as Grand River Aseptic Manufacturing, Pyramid Bioservices, and PCI, which offer a narrower range of services (e.g., exclusively drug substance or drug product). Lastly, organizations such as Pfizer CentreOne and AbbVie Contract Manufacturing, which offer a portfolio of manufacturing services to increase utilization of internal manufacturing capabilities. In some instances, we also compete with in-house research, development and manufacturing departments of other biopharmaceutical companies.
MANUFACTURING OPERATIONS
Manufacturing Network: Emergent relies on an internal and external network of manufacturers and other third parties to produce and test its commercial and clinical supply of products. The following products are manufactured internally: ACAM2000®, ANTHRASIL®, BAT® and CNJ-016® (VIGIV).
NARCAN® Nasal Spray, EBANGA™, TEMBEXA®, CYFENDUS® and BioThrax® are produced externally in whole or in part by contract manufacturers. However, we perform the majority of the work internally to produce CYFENDUS® and BioThrax®.
For example, materials for production of NARCAN® Nasal Spray, such as the naloxone active pharmaceutical ingredient and other excipients, along with the vial, stopper and device are produced around the world by other third parties and delivered to the primary manufacturer and released to manufacturing following appropriate testing.
15
Supplies and Raw Materials: We place purchase orders for quantities of raw material and supplies in advance of their use in manufacturing in quantities believed to be sufficient to meet upcoming demand requirements. Once received into our facilities or our contract manufacturer’s facility, these materials are subject to rigorous testing to ensure they are appropriate for use.
We obtain Alhydrogel® adjuvant 2%, which is used in the manufacture of CYFENDUS® and BioThrax® vaccines, from a single-source supplier for which we currently have no alternative source of supply. However, we maintain stored supplies of this adjuvant in quantities we believe to be sufficient to meet our expected manufacturing needs. We also utilize single-source suppliers for other raw materials in our manufacturing processes.
In addition, we utilize single source suppliers for all components of NARCAN® Nasal Spray.
We also rely on single source suppliers for our plasma collection to support the CNJ-016® (VIGIV) and BAT® programs. We work closely with our suppliers for these specialty programs and operate under long-term agreements.
INTELLECTUAL PROPERTY
We actively seek to protect intellectual property related to our assets, including patent rights, trademark rights, trade secrets and proprietary confidential information, through defense and enforcement of existing rights and pursuit of protection on new and arising innovations. The duration of and the type of protection for patent rights depends upon many factors including the type of patent, the scope of its coverage, the availability of regulatory-related extensions or administrative term adjustments, the availability of legal remedies in a particular country, and the validity and enforceability of the patents. We are a party to various license agreements, including those under which we license patents, patent applications, trademarks, know-how, and other intellectual property rights. It is our policy to ethically consider the enforcement and defense of our intellectual property rights, and to respect the valid and enforceable intellectual property rights of others.
REGULATION
Regulations in the U.S. and other countries have a significant impact on our product development, manufacturing and marketing activities.
Government Contracting
Our status as a USG contractor means that we are subject to various statutes and regulations, including:
•the Federal Acquisition Regulation ("FAR") and agency-specific regulations supplemental to FAR, which comprehensively regulate the award, formation, administration and performance of government contracts;
•the Defense Federal Acquisition Regulations Supplement ("DFARS") and agency-specific regulations supplemental to DFARS, which comprehensively regulate the award, formation, administration and performance of DoW government contracts;
•the Department of State Acquisition Regulation which regulates the relationship between a Department of State organization and a contractor or potential contractor;
•business ethics and public integrity obligations, which govern conflicts of interest and the hiring of former government employees, restrict the granting of gratuities and funding of lobbying activities and incorporate other requirements such as the Anti-Kickback Act, the Procurement Integrity Act, the False Claims Act and the Foreign Corrupt Practices Act;
•export and import control laws and regulations, including but not limited to the Export Administration Regulations and International Traffic in Arms Regulations; and
•laws, regulations and executive orders restricting the use and dissemination of information classified for national security purposes and the exportation of certain products and technical data.
USG agencies routinely audit and investigate government contractors for compliance with applicable laws and standards. Our role and status as a large government supplier to HHS, particularly BARDA increases the likelihood of Congressional review and oversight. The legal framework we are subject to as a government contractor imposes stricter penalties than those normally applicable to commercial contracts, such as criminal and civil liability and suspension and debarment from future government contracting. In addition, pursuant to various laws, our government contracts can be subject to unilateral termination or modification by the government for convenience, detailed auditing and accounting systems requirements, statutorily controlled pricing, sourcing and subcontracting restrictions and statutorily mandated processes for adjudicating contract disputes.
16
The Project BioShield Act of 2004. The Project BioShield Act of 2004 (Project BioShield) was enacted to augment market incentives for companies pursuing the development of MCMs of which the government is the only significant market. Project BioShield provided $5.6 billion over 10 years to develop, purchase, and stockpile MCMs for use in a public health emergency against CBRNE agents.
The Pandemic and All Hazards Preparedness Act of 2006 and Reauthorization Acts. The Pandemic and All Hazards Preparedness Act of 2006 established the role of ASPR within HHS and provided statutory authorities for a number of programs, including the creation of BARDA to support the development and procurement of MCMs to respond to CBRNE. The Pandemic All Hazards Preparedness Reauthorization Act of 2013 ("PAHPRA") continued BARDA’s role and reauthorized Project BioShield funding through fiscal year 2018 and provided BARDA with additional appropriations to support advanced research and development. The Pandemic and All-Hazards Preparedness and Advancing Innovation Act of 2019 reauthorized Project BioShield’s special reserve fund and authorized 10-year funding for product development. BARDA has used the incentives under Project BioShield and subsequent reauthorizations of it to build a robust pipeline of MCMs for multiple CBRNE agents. It has also procured and stockpiled many of our related products for potential use in the event of a PHT emergency, including BioThrax®, ACAM2000®, ANTHRASIL®, BAT®, CNJ-016® (VIGIV) and raxibacumab products.
Funding for BARDA is provided by annual appropriations by Congress. Congress appropriates annual funding for procurement of MCMs for the SNS (currently managed by ASPR) and for the NIAID to conduct biodefense research. This appropriation funding supplements amounts available under Project BioShield.
Emergency Use Authorization
Section 564 of the Federal Food, Drug, and Cosmetics Act ("FDCA") authorizes FDA to issue an Emergency Use Authorization ("EUA") to permit the introduction into interstate commerce of unapproved MCMs, or approved MCMs for unapproved uses, in the context of certain potential or actual public health emergencies. Several actions are required to trigger FDA’s authority to issue EUAs. First, there must be a determination by certain federal officials that a particular threat or emergency exists. This can be (1) a determination by the Secretary of HHS that there is a public health emergency, or a significant potential for a public health emergency, that affects, or has a significant potential to affect, national security or the health and security of United States citizens living abroad, and that involves CBRN agents, or a disease or condition that may be attributable to CBRN agents; (2) a determination by the Secretary of Homeland Security (“DHS”) that there is a domestic emergency, or a significant potential for a domestic emergency, involving a heightened risk of attack with a CBRN agent; (3) a determination by the Secretary of War that there is a military emergency, or a significant potential for a military emergency, involving a heightened risk to United States military forces from an attack with a CBRN agent or an agent that may cause, or is otherwise associated with, an imminently life-threatening and specific risk to United States military forces; or (4) the identification of a material threat pursuant to section 319F–2 of the Public Health Service Act (“PHSA”) sufficient to affect national security or the health and security of United States citizens living abroad. Based on one of these determinations, the Secretary of HHS may make a declaration (the EUA Declaration) that circumstances exist justifying EUAs for MCMs to respond to the threat or emergency at issue. Once the relevant determination and EUA declaration are issued, FDA has the authority to issue EUAs for the use of specific medical products based on criteria established by statute, including that the product at issue may be effective in diagnosing, treating, or preventing a serious or life-threatening disease or condition related to the threat or emergency and that there are no adequate, approved, and available alternatives to the product for diagnosing, preventing, or treating the disease or condition. EUAs are subject to additional conditions and restrictions, are product-specific, and terminate when the EUA is revoked or the EUA declaration is terminated because the Secretary of HHS has determined that the circumstances that led to the emergency determination have ceased or because the authorized use has been approved.
Under PAHPRA, the USG may purchase certain MCMs for the SNS prior to FDA approval, licensure or authorization, under certain circumstances. BARDA is currently procuring CYFENDUS® (Anthrax Vaccine Adsorbed (AVA), Adjuvanted).
Public Readiness and Emergency Preparedness Act. The Public Readiness and Emergency Preparedness Act ("PREP Act") creates certain liability protections for manufacturers of MCMs when the Secretary of HHS issues a declaration related to a specific disease, condition or public health threat. The PREP Act is intended to provide liability immunity from claims under federal or state law for loss caused by, arising out of, relating to, or resulting from the administration or use of a covered MCM. The scope of protection will be defined by the specific PREP Act declaration. The only statutory exception to the immunity granted by the PREP Act is for actions or failures to act that constitute willful misconduct. The Secretary of HHS has issued PREP Act declarations covering MCMs for smallpox, mpox, and other orthopox; anthrax; and botulinum toxin, among other pathogens. These declarations could apply to ACAM2000®, ANTHRASIL®, BAT®, BioThrax®, CYFENDUS®, raxibacumab and CNJ-016® (VIGIV) products, as covered MCMs. The declarations for anthrax and botulism expire on December 31, 2027. The declaration for smallpox, mpox, and other orthopox expires on December 31, 2032.
17
Support Anti-Terrorism by Fostering Effective Technology Act of 2002. The Support Anti-terrorism by Fostering Effective Technologies Act of 2002 (“SAFETY Act”) was enacted to create certain liability limitations for Qualified Anti-Terrorism Technologies (“QATTs”) for claims arising out of, related to, or resulting from an act of terrorism. DHS administers the SAFETY Act program, which provides two potential categories of liability protections – designation and certification. If DHS deems an MCM a “Designated Technology,” then the company’s liability is limited to the amount of liability insurance that DHS determines the company must maintain. To receive “certification,” a QATT must first be “designated” and also be shown to perform as intended, conform to the manufacturer’s specifications, and be safe for use as intended. Certification allows the company to assert the Government Contractor defense for claims arising from acts of terrorism.
DHS granted SAFETY Act designation and certification for BioThrax® in 2006 and has continued to renew those determinations. Any future renewals of the SAFETY Act designation and certification for BioThrax® products may not provide adequate protection from all claims made against us.
Product Development for Therapeutics and Vaccines
Pre-Clinical Testing. We generally perform pre-clinical safety and efficacy testing on our product candidates before we initiate clinical trials.
Animal Rule. Conducting controlled human clinical trials to determine efficacy of MCMs against dangerous pathogens may sometimes be unethical or unfeasible. In such circumstances, products may be approved under the FDA’s “Animal Rule.” According to the FDA, this regulatory pathway can only be pursued if conducting human efficacy studies would be unethical and field trials to study the product’s effectiveness, after an accidental or deliberate exposure, are not feasible. Under the “Animal Rule,” under some circumstances, approval of product candidates can be based on efficacy data from animal studies. In assessing the sufficiency of animal data, the FDA may take into account other available data, including human data. These approvals generally are associated with a requirement for post-approval trials that would be conducted in the event of an act of bioterrorism, a pandemic, or other natural exposure to the pathogen at issue.
Investigational New Drug Application. Before clinical testing may begin, the results of pre-clinical testing and other available clinical data and manufacturing information must be submitted to the FDA as part of an Investigational New Drug application ("IND"). The data must provide an adequate basis for evaluating both the safety and the scientific rationale for the initial clinical studies. The FDA may impose a full or partial clinical hold on the effectiveness of an IND pending receipt of additional information.
Clinical Trials. Clinical trials involve administration of a product candidate to healthy human volunteers or patients under the supervision of a qualified physician under a regulatory agency approved protocol for the country in which the human trial is to be conducted. Human clinical trials typically are conducted in the following three sequential phases.
▪Phase 1 studies involve introduction of the drug into human subjects (usually healthy, but in some circumstances may include patients) to assess safety, metabolism, pharmacokinetics, pharmacological actions, side effects and early evidence of effectiveness.
▪Phase 2 involves studies to assess the efficacy of the drug in specific, targeted indications to explore preliminary efficacy, tolerance, optimal dosage, and safety.
▪Phase 3 involves pivotal trials to assess clinical efficacy and safety in a larger number of healthy subjects or patients. These trials are intended to permit the FDA to evaluate the overall benefit-risk relationship of the product, and provide adequate information for approval and drug labeling.
In addition, in certain circumstances Phase 4 studies may be conducted following marketing approval in order to provide additional data related to drug use. The FDA may impose a temporary or permanent clinical hold, or other sanctions, if it believes that a clinical trial is not being conducted in accordance with the FDA requirements or presents an unacceptable risk to the clinical trial subjects.
Good Clinical Practice and International Conference on Harmonization ("ICH") Guidelines. All phases of clinical studies must be conducted in conformance with the FDA’s bioresearch monitoring regulations and Good Clinical Practices ("GCP"), which include applicable ICH Guidelines and are collectively ethical and scientific quality standards for the conduct and reporting of clinical trials in human subjects.
18
Marketing Approval – Biologics, Drugs and Vaccines
Biologics License Application ("BLA")/New Drug Application ("NDA"). For large molecule products, such as vaccines, products derived from blood and blood components, and antibodies, all data obtained from a development program, including research and product development, manufacturing, pre-clinical and clinical trials, labeling and related information are submitted in a BLA to the FDA and in similar regulatory filings with the corresponding agencies in other countries for review and approval. For small molecule drugs, this information is submitted in an NDA filing. The submission of an application, either a BLA or an NDA, is not a guarantee that the FDA will find the application complete and accept it for filing. The FDA may issue a refuse to file, or RTF, letter to the applicant and request additional information, in which case the application must be resubmitted. Most applications are subject to a substantial application fee and each approved product will be assessed an annual fee. Under the FDCA, the FDA has the authority to grant waivers of certain user fees.
In reviewing a BLA or NDA, the FDA may grant approval, request more information or data, or decline to approve the application if, among other potential deficiencies, the FDA determines that the application does not provide substantial evidence of effectiveness, the drug is not safe for use under the conditions of use in the proposed labeling, or there are deficiencies in manufacturing quality. If the FDA decides not to approve an application, it will issue a complete response letter, or CRL. During the FDA's review of the application, the FDA will also typically inspect one or more clinical sites to ensure compliance with GCPs as well as the facility or facilities at which the candidate is manufactured to ensure compliance with current good manufacturing practices ("CGMPs").
The receipt of regulatory approval may take many years, and typically involves the expenditure of substantial financial resources. The FDA may also impose conditions upon approval or significantly limit the indications approved for a given product and/or require, as a condition of approval, enhanced labeling, packaging, post-approval clinical trials, expedited reporting of certain adverse events, pre-approval of promotional materials or restrictions on consumer advertising, which could negatively impact the commercial success of a product.
Abbreviated New Drug Applications Under Section 505(j) and Section 505(b)(2) New Drug Applications. Most drug products obtain FDA marketing approval under a full NDA for innovator products, or an abbreviated new drug application ("ANDA") for generic products. The Hatch-Waxman amendments to the FDCA established a statutory procedure for submission and FDA review and approval of ANDAs for generic versions of branded drugs previously approved by the FDA (reference listed drugs, or RLDs). Because the safety and efficacy of RLDs have already been established by the brand company (sometimes referred to as the innovator), the FDA does not generally require ANDA applicants to provide preclinical and full clinical data to establish safety and effectiveness. However, a generic manufacturer is required to demonstrate bioequivalence (i.e., it provides the equivalent rate and extent of absorption as the innovator product) and meet other requirements (including that the generic product provides the same active ingredient in the same dose and dosage form via the same route of administration).
A third alternative for approval of a drug product is commonly referred to as a Section 505(b)(2) NDA, which enables the applicant to rely, in part, on the FDA’s findings of safety and efficacy of an existing product in support of its application. Section 505(b)(2) NDAs often provide an alternate path to FDA approval for new or improved formulations, new strengths, new routes of administration or new uses of previously approved products. Section 505(b)(2) permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. The applicant might rely upon the FDA’s findings with respect to certain pre-clinical or clinical studies conducted for an approved product. The FDA may also require companies to perform additional studies or submit other information to support the change from the approved product. The FDA may then approve the new product candidate for certain indications for which the referenced product has been approved, as well as for any new indication sought by the applicant.
In seeking approval for a drug through an NDA, including a 505(b)(2) NDA, applicants are required to submit to the FDA information about certain patents of the applicant or that are held by third parties whose claims cover the applicant’s product. Upon approval of an NDA, each of the patents for which the applicant has submitted information in connection with the NDA is then published in the Orange Book. Any subsequent applicant who files an ANDA or a 505(b)(2) NDA must make one of the following certifications to the FDA concerning each patent for which the RLD sponsor was required to submit information in connection with the RLD: (1) the patent information has not been submitted to the FDA; (2) the patent has expired; (3) the date on which the patent will expire; or (4) the patent is invalid, unenforceable, or will not be infringed by the manufacture, use or sale of the drug product for which the application is submitted. This last certification is known as a paragraph IV certification. Alternatively, the ANDA or 505(b)(2) NDA applicant may submit a statement that there are no relevant patents or that a method-of-use patent does not claim a proposed indication or other condition of use for which the applicant is seeking approval.
19
If the RLD’s NDA holder or patent owner initiates patent litigation to enforce an Orange Book-listed patent within 45 days after receiving notice of a paragraph IV certification, the FDA generally is prohibited from approving the application until 30 months from the date of receipt of the paragraph IV notice, although this stay may terminate earlier depending upon the resolution of the litigation, if the court issues an order terminating the stay, or if the patent owner or exclusive patent licensee consents to approval of the application before the expiration of the stay. The ANDA or 505(b)(2) application also will not be approved until any applicable non-patent exclusivity listed in the Orange Book for the RLD has expired.
Biosimilar Products. When a biological product is licensed for marketing by FDA through the approval of a BLA under section 351(a) of the PHSA, the product may be entitled to exclusivity barring FDA from accepting or approving an application under section 351(k) of the PHSA for a competing product for certain periods of time. The Biologics Price Competition and Innovation Act of 2009 (the "BPCIA") added Section 351(k) of the PHSA, which provides an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference product. The FDA may approve a biosimilar product if it finds that the product is highly similar to the reference product notwithstanding minor differences in clinically inactive components and there are no clinically meaningful differences between the proposed biosimilar product and the reference product in terms of safety, purity, and potency. For the FDA to approve an interchangeable biosimilar product, it must conclude that the product is biosimilar to the reference product, can be expected to produce the same clinical result as the reference product in any given patient, and—for a product that is administered more than once to an individual—alternating or switching between the proposed interchangeable product and the reference product would not create an increased risk in terms of safety or diminished efficacy compared to using the reference product only.
FDA will not accept a biosimilar application until four years after the date of first licensure of a biological product licensed under section 351(a) of the PHSA, and FDA will not approve a biosimilar application until 12 years after such date of first licensure. This type of exclusivity is known as reference product exclusivity. The approval of a supplemental BLA or certain subsequent BLAs does not give rise to a new date of first licensure, and, consequently, does not yield an additional period of reference product exclusivity. Moreover, reference product exclusivity does not affect the timing of FDA’s acceptance or approval of a competing sponsor’s section 351(a) BLA containing the sponsor’s own pre-clinical data and data from adequate and well‑controlled clinical trials to demonstrate the safety, purity, and potency of its product. There have been legislative proposals to reduce the duration of the exclusivity periods, but none has been enacted to date. Moreover, many states have enacted laws that address pharmacy substitution practices involving biosimilar products.
Post-Approval Requirements. Any drug, biologic or medical device product for which we receive FDA marketing authorization will be subject to continuing regulation by the FDA, including, among other things, record keeping requirements, reporting of adverse events, providing FDA with updated safety and efficacy information, filing changes to manufacturing process and analytical testing, product sampling and distribution requirements (for drugs and biologics), restrictions on advertising and promotion, and FDA inspections. Adverse events that are reported after marketing approval can result in additional limitations being placed on the product’s distribution or use and, potentially, withdrawal or suspension of the product from the market. The FDA may also require post-approval clinical trials and/or labeling changes.
Facilities involved in the manufacture and distribution of approved products are required to be registered with the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA for compliance with CGMP and other laws.
A company that is found to have improperly promoted unapproved or off-label uses or otherwise not to have met applicable promotion rules may be subject to significant liability under both the FDCA and other statutes, including the False Claims Act.
Orphan Drugs. Under the Orphan Drug Act, an applicant can request the FDA to designate a product as an “orphan drug" in the U.S. if the drug is intended to treat a rare disease or condition. A disease or condition is considered rare if it affects fewer than 200,000 people in the U.S. or there is no reasonable expectation that the cost of developing the drug and making it available in the United States will be recovered from sales in the United States. A manufacturer must request orphan drug designation prior to submitting a BLA or NDA. Products designated as orphan drugs may be eligible for special grant funding for R&D, FDA assistance with the review of clinical trial protocols, potential tax credits for research, an exemption from the application fee for marketing applications and potentially a seven-year period of orphan drug exclusivity after marketing approval. A grant of an orphan designation is not a guarantee that a product will be approved.
20
Orphan drug exclusivity (afforded to the first applicant to receive approval for an orphan designated drug for a particular rare disease or condition) generally prevents FDA approval of another sponsor’s application for the same drug for the same indication for seven years after approval of the product that received such exclusivity unless the Orphan drug exclusivity will not bar approval of the same drug marketed by a different manufacturer under certain circumstances, including if the company with orphan drug exclusivity is not able to meet market demand, grants consent to the FDA’s approval of the subsequent product or the subsequent product is shown to be clinically superior to the approved product on the basis of greater efficacy or safety, or providing a major contribution to patient care.
After an appellate court decision issued in 2021 called into question the continued validity of FDA's approach to determining the scope of orphan-drug exclusivity, the FDA announced in January 2023 that it would continue to apply the FDA’s regulations limiting the scope of orphan drug exclusivity to a product’s approved uses or indications. As a result of the FDA’s announcement, the scope of orphan drug exclusivity and other issues relating to the FDA’s implementation of the Orphan Drug Act with respect to previously approved and future products may be the subject of litigation or legislation.
Vaccine and Therapeutic Product Lot Release Requirements. The manufacturing process for biological products is complex, therefore the FDA requires for many biologics, including most vaccines and immune globulin products, that each product lot undergo FDA testing for purity, potency, identity and sterility. FDA may request samples of any lot and, when deemed necessary for the safety, purity, and potency of the product, FDA may prohibit us from distributing a lot until FDA releases the lot. Some of our vaccines are subject to lot release protocols by the FDA and other regulatory agencies.
Marketing Approval – Devices
Devices may be marketed as stand-alone devices or as constituent parts of a Combination Product, such as a device for delivery of a drug product. Unless an exemption applies, each medical device commercially distributed in the United States requires either FDA clearance of a 510(k) premarket notification, approval of a premarket approval application (“PMA”) or issuance of a de novo classification order.
Medical devices are classified into one of three classes—Class I, Class II or Class III—depending on the degree of risk and the level of control necessary to assure the safety and effectiveness of each medical device. Medical devices deemed to pose lower risks are generally placed in either Class I or II. While most Class I devices are exempt from the 510(k) premarket notification requirement, manufacturers of most Class II devices are required to submit to the FDA a pre-market notification. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining life-supporting or many implantable devices, or devices that have been found not substantially equivalent to a legally marketed Class I or Class II predicate device, are placed in Class III, requiring approval of a PMA.
All clinical investigations of devices to determine safety and effectiveness must be conducted in accordance with the FDA’s investigational device exemption (“IDE”) regulations that govern investigational device labeling, prohibit promotion of the investigational device, and specify an array of study review and approval, informed consent, recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. If the device presents a “significant risk” to human health, as defined by the FDA, the FDA requires the device sponsor to submit an IDE application to the FDA, which must become effective prior to commencing human clinical trials. All clinical device studies, including non-significant risk studies, must be approved by, and conducted under the oversight of, an Institutional Review Board (“IRB”). The IRB is responsible for the initial and continuing review of the study and may pose additional requirements for the conduct of the study.
Both before and after a medical device is commercially distributed, manufacturers and marketers of the device have ongoing responsibilities under FDA regulations, including, for example, establishment registration and device listing; compliance with the requirements of the Quality Management System Regulation (“QMSR”); compliance with requirements regarding the labeling and marketing of devices; medical device reporting regulations; correction and removal reporting regulations; compliance with requirements for Unique Device Identification (“UDI”); and post-market surveillance activities and requirements.
Device manufacturers are subject to periodic and unannounced inspection by the FDA. The FDA reviews design and manufacturing practices, record keeping, reports of adverse events, labeling and other information to ensure compliance with the QMSR and other applicable requirements, and to identify potential problems with manufacturing processes and marketed medical devices.
21
A combination product is a product comprised of two or more regulated components (e.g., a drug and device) that are combined into a single product, co-packaged, or sold separately but intended for co-administration, as evidenced by the labeling for the products (cross-labeling). Like their constituent parts—e.g., drugs and devices—combination products are highly regulated and subject to a broad range of pre- and post-market requirements including premarket review, CGMPs and/or QMSRs, adverse event reporting, periodic reports, labeling and advertising and promotion requirements and restrictions, market withdrawal and recall. When issuing the QMSR, the FDA made conforming edits to the combination product regulation to clarify the device Quality Management System ("QMS") requirements for combination products. Combination products are typically reviewed through a marketing submission that corresponds to the constituent part which provides the primary mode of action (“PMOA”) for the combination product. For example, if the PMOA of a device-biologic combination product is attributable to the biologic, the agency center that reviews biologics would have the primary jurisdiction for the review.
The FDA also regulates the export of medical devices from the U.S., and medical devices that are not authorized to be legally marketed in the U.S. are subject to FDA export requirements.
Manufacturing Requirements
The FDA's statutory provisions and regulations require that drugs be manufactured in FDA-registered facilities and in accordance with CGMPs. The CGMP regulations include requirements relating to organization and personnel, buildings and facilities, equipment, control of components and product containers and closures, production and process controls, packaging and labeling controls, holding and distribution, laboratory controls, records and reports, and returned and salvaged products. These processes must be performed in compliance with the applicable portions of the QMSR, which covers the methods and the facilities and controls for the manufacture, testing, production, quality assurance, labeling, packaging, distribution, installation and servicing of finished devices intended for human use. Manufacturers and other entities involved in the manufacture and distribution of cleared, approved, or otherwise authorized products are required to register their establishments with the FDA, and in some instances state agencies, and they are subject to periodic unannounced or announced inspections by the FDA for compliance with CGMPs and/or QMSRs and other requirements.
The FDA uses a "risk-based approach" to inspections, whereby the agency prioritizes medical product surveillance inspections deemed high risk based on a variety of specific criteria, such as facility type, compliance history, and inherent risks of product manufactured at the facility. Manufacturers may also have to provide, on request, electronic or physical records regarding their establishments. Delaying, denying, limiting, or refusing inspection by the FDA may lead to a product being deemed to be adulterated. Changes to the manufacturing process, specifications or container closure system for an approved drug product are strictly regulated and often require prior FDA approval before being implemented. The FDA's regulations also require, among other things, the investigation and correction of any deviations from CGMP or failures to follow the QMSR and the maintenance of applicable documentation by the sponsor and any third-party manufacturers involved in producing the approved, cleared, or otherwise authorized product.
Regulation Outside of the U.S.
Currently, we maintain a commercial presence in the U.S. and Canada as well as certain other countries. Each foreign country has its own regulatory requirements for medicines and medical devices. In the EU, medicinal products are authorized following a process that is similarly demanding as the process required in the U.S. Drug products may be authorized in one of two ways, either through the mutual recognition/decentralized procedure, which provides for the mutual recognition procedure of national approval decisions by the competent authorities of the EU Member States or through the centralized procedure, which provides for the grant of a single marketing authorization that is valid for all EU member states. Before a new medical device can be placed on the market in the EU, compliance with the requirements of the Medical Devices Regulation (EU) 2017/745 must be demonstrated in order to affix the CE Mark to the product. The method of assessing conformity varies depending on the class of the product, but normally involves a combination of self-assessment by the manufacturer and a third-party assessment by a notified body. We are also subject to many of the same continuing post-approval requirements in the EU as we are in the U.S. (e.g., good manufacturing practices).
The Medicines and Healthcare Products Regulatory Agency (the “MHRA”) serves as the regulatory authority overseeing medicines and medical devices in the United Kingdom.
Additionally, an International Recognition Procedure (the “IRP”) took effect in January 2024, designed to facilitate marketing authorizations in the UK for products already approved by a recognized regulatory authority. The IRP applies to products authorized by the European Medicines Agency (for EU centralized approvals), national regulators in the European Economic Area (the “EEA”), and the FDA, among others. However, reliance-based or recognition-based regulatory decisions are not eligible under this procedure.
22
Potential Sanctions
For all FDA-regulated products, if the FDA finds that a manufacturer has failed to comply with applicable laws and regulations, or that a product is ineffective or poses an unreasonable health risk, it can institute or seek a wide variety of enforcement actions and remedies, including but not limited to:
•restrictions on products, manufacturers or manufacturing processes;
•restrictions on the labeling or marketing of a product;
•restrictions on distribution or use of a product;
•requirements to conduct post-marketing studies or clinical trials;
•warning letters or untitled letters;
•withdrawal of the products from the market;
•refusal to approve pending applications or supplements to approved applications that are submitted;
•recall of products;
•fines, restitution or disgorgement of profits or revenues;
•suspension or withdrawal of marketing approvals;
•refusal to permit the import or export of our products;
•product seizure; and
•injunctions or the imposition of civil or criminal penalties.
Health regulatory authorities in other countries have similar rules and regulations although the specifics vary from jurisdiction to jurisdiction.
Fraud, Abuse and Anti-Corruption Laws
The U.S. and most other jurisdictions have detailed requirements that apply to government and private health care programs, and a broad range of fraud and abuse laws, transparency laws, and other laws. Relevant U.S. federal and state healthcare laws and regulations include:
•The federal Anti-Kickback Statute;
•The False Claims Act;
•The federal Health Insurance Portability and Accountability Act of 1996 ("HIPAA"), as amended by the Health Information Technology for Economic and Clinical Health ("HITECH") Act;
•The price reporting requirements under the Medicaid Drug Rebate Program and the Veterans Health Care Act of 1992;
•The federal Physician Payment Sunshine Act, being implemented as the Open Payments Program; and
•Analogous and similar state laws and regulations.
Our operations are also subject to compliance with the Foreign Corrupt Practices Act ("FCPA") which prohibits corporations and individuals from corruptly paying, offering to pay, or authorizing the payment of anything of value to any foreign government official, government staff member, political party or party official, or political candidate, directly or indirectly, in an attempt to influence a person working in an official capacity or otherwise obtain an improper advantage. We also may be impacted under the FCPA by the activities of our distributors, collaborators, contract research organizations, vendors, consultants, agents, or other business partners. As a public company, the FCPA also requires us to make and keep books and records that accurately and fairly reflect all of our transactions and to devise and maintain an adequate system of internal accounting controls. Our operations are also subject to compliance with the U.K. Bribery Act, which applies to bribery activities both in the public and private sector, Canada’s Corruption of Foreign Public Officials Act and similar laws in other countries.
We have a global corporate compliance program designed to identify, prevent, and mitigate healthcare fraud and abuse risk through, among other things, policies, systems and promoting a culture of compliance. Our failure to comply with these laws and regulations could subject us to criminal or civil penalties.
23
Regulations Governing Reimbursement
The marketing practices of U.S. pharmaceutical manufacturers are also subject to federal and state healthcare laws related to government funded healthcare programs.
In the U.S., certain of our products are reimbursed under federal and state health care programs such as Medicaid, Medicare, Tricare, and/or state pharmaceutical assistance programs. Many foreign countries have similar laws.
Various U.S. federal health care laws apply when we or customers submit claims for items or services that are reimbursed under federally funded health care programs, including federal and state anti-kickback laws, false claims laws, and anti-self-referral laws, which may apply to federal and state-funded health care programs and private third-party payers.
Failure to comply with these laws and regulations could subject us to criminal or civil penalties.
Additionally, drug pricing is an active area for regulatory reform at the federal and state levels, and significant changes to current drug pricing and reimbursement structures in the U.S. continue to be considered and enacted. For example, the Inflation Reduction Act of 2022 (the “IRA”), among other provisions, gives HHS the ability and authority to directly negotiate with manufacturers the price that Medicare will pay for certain single-source drugs that account for high Medicare spending. The IRA will also require manufacturers of certain Part B and Part D drugs to pay to HHS rebates based on certain calculations and triggers (i.e., when drug prices increase and outpace the rate of inflation). The Medicare Drug Price Negotiation Program may affect future Medicare reimbursement for certain of our products.
Data Privacy Laws
A number of U.S. states have passed or introduced comprehensive privacy laws that impose operational requirements on companies processing personal data, similar in many respects to the General Data Protection Regulation (“GDPR”) in the European Union (“State Consumer Privacy Laws”). Canada’s Personal Information Protection and Electronic Documents Act (“PIPEDA”) also establishes a federal privacy framework requiring organizations to obtain meaningful consent, limit data use to identified purposes, and implement safeguards for personal information. While PIPEDA differs from U.S. state laws in scope and enforcement, it reflects similar principles of transparency, accountability, and consumer rights. As of December 31, 2025, comprehensive State Consumer Privacy Laws were effective across a growing number of U.S. states—including California, Colorado, Virginia, Connecticut, Utah, Florida, Texas, Oregon, and Montana—with additional enacted laws in Iowa, Delaware, Nebraska, New Hampshire, New Jersey, Tennessee, Minnesota, and Maryland, and further laws in Rhode Island, Kentucky, and Indiana becoming effective in 2025 and 2026. These state laws generally apply based on the processing of personal data of state residents, provided that certain other conditions are met and the Company does business in the relevant state. As a result, these laws may, in some cases, apply to companies that do not maintain physical operations in the state.
As an example of these State Consumer Privacy Laws, the California Consumer Privacy Act of 2018 (“CCPA”), which came into effect in January 2020, requires covered companies processing personal information of California residents to make disclosures regarding their data collection, use, and sharing practices, provides consumers with the ability to opt out of certain data sharing with third parties, places restrictions on the use of dark patterns in certain circumstances, and establishes a private right of action for certain data breaches. As states continue to introduce and expand comprehensive privacy frameworks, several have also begun addressing emerging issues associated with automated decision‑making and AI‑enabled processing. While our use of AI remains limited and in early stages, some state privacy laws include requirements governing profiling, risk assessments, and transparency related to automated data decision making. These developments underscore the evolving regulatory expectations around responsible AI use and the corresponding privacy obligations for companies handling consumer data.
Certain states, such as Nevada and Washington, have recently enacted consumer health data laws for the protection of consumers' personal health data by outlining broad definitions of consumer health data, a range of consumer rights and valid consent and other compliance requirements. Although similar to State Consumer Privacy Laws, these consumer health data laws impose restrictions and obligations beyond State Consumer Privacy Laws.
Additionally, there are a number of other federal, state, and international legal frameworks that bear on the collection use, dissemination and security of data. Among them, the Federal Trade Commission and many state attorneys general are interpreting federal and state consumer protection laws to impose standards for the online collection, use, dissemination and security of data and there are federal and state wiretap laws that may be relevant to data collection and sharing. In addition, the U.S. Department of Justice (“DOJ”) final rule implementing Executive Order 14117 (“Preventing Access to American’s Bulk Sensitive Personal Data and United States Government-Related Data by Countries of Concern”) has taken effect, and it prohibits or restricts certain transactions involving access by “countries of concern” or “covered persons” to “government-related data” or “bulk U.S. sensitive personal data.” The final rule imposes certain diligence, security, and audit record-keeping obligations, among other requirements.
24
Several states have recently emphasized requirements related to consumer consent flows, including guidelines regarding cookie banner options, restrictions on dark patterns, and obligations to honor browser‑based opt‑out signals. We have continued to take steps to reflect these requirements across our consumer‑facing sites.
The compliance and other burdens imposed by the EU's GDPR, CCPA and other privacy laws and regulations may be substantial as they are subject to differing interpretations and implementation among jurisdictions. The restrictions imposed by such laws may require us to modify our data handling practices and impose additional compliance costs and burdens.
Other Industry Regulation
Our present and future business has been and will continue to be subject to various other laws and regulations. Various laws, regulations and recommendations relating to safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import, export, use and disposal of hazardous or potentially hazardous substances, including radioactive compounds and infectious disease agents used in connection with our product development, are or may be applicable to our activities.
HUMAN CAPITAL
We value our employees and the contributions each of them makes to achieving our mission to protect and save lives. We strive to create an environment that is professionally and personally rewarding by offering challenging work and projects for individual and team contribution, and opportunities for professional and personal development. Our commitment to attracting, developing, and retaining talented employees helps us to fuel our business growth, drive innovation in the products and services we develop, and inform the way we solve problems and how we serve the needs of a global and diverse patient, customer and partner base. We believe that our investments in employee engagement and leadership development are essential to building the capabilities needed to realize our business strategy.
As of December 31, 2025, we had approximately 900 employees globally. During 2025, our number of employees globally stayed consistent.
We focus on building leaders at every level with the requisite scientific, technical and professional skills to develop and deliver products and services that protect life. We have consistent talent processes and systems across the company including performance management, training and development and succession planning. We recognize the need for ongoing skill enhancement and support continued learning through on-the-job assignments, leadership and technical training programs, tuition assistance professional memberships and professional conference attendance. We continue to use the Gallup Q12 employee engagement survey and administer “pulse surveys” throughout the year to gather feedback on matters of interest and importance to our employees and our business.
Our total rewards plan consists of comprehensive benefits and salaries, bonuses, and for employees in eligible roles, equity awards. In addition, in 2025 we introduced an "equity for all" award that included an equity grant for employees not typically eligible for equity. We continue to provide employees access to country-specific salary range information so that they may have greater visibility to their current compensation levels and more context as they explore developing their careers within our company.
In addition, we are deeply committed to the health and well-being of our employees. We provide a comprehensive range of benefits and resources to support their physical, mental, and financial wellness. Additionally, our safety programs comply with regional requirements and are designed to ensure a safe and comfortable work environment, including a strong culture of safety in our day-to-day operations.
SUSTAINABILITY AND CORPORATE RESPONSIBILITY
Our mission to protect and save lives motivates us to explore our impact at a broader scale. Our approach to these issues is the foundation of good governance and strengthens accountability in all aspects of our business activities and relationships. In 2025, we completed a sustainability compliance roadmap to help us navigate and anticipate reporting expectations and requirements within the countries we operate and to establish sustainability and reporting priorities. In addition, for the first time, we completed the limited assurance process for our 2024 Scope 1 and 2 GHG emissions and received an “Independent Verification Opinion Declaration” from our auditors. In 2026, we will continue to monitor and prepare for emerging sustainability regulations globally, which includes completing a Double Materiality Assessment (DMA).
Our approach is influenced by the framework developed by the Task Force on Climate-Related Disclosures as well as the Sustainability Accounting Standards Board’s standards focused on the healthcare, biotechnology, and pharmaceutical industries. The SASB standards provide guidelines on key sustainability issues that directly impact the operational performance and financial condition of our company.
25
Priority Issues
Our priority issues are as follows:
•Top Priorities
◦Talent Attraction, Engagement & Development
◦Ethics & Compliance
◦Product Quality & Patient Safety
◦Sustainable Innovation
◦Product Affordability & Accessibility
◦Responsible Supply Chain
•Relative Priorities
◦Supplier Product Quality, Reliability and Compliance
◦Clinical Trial Practices
◦Employee Health and Safety
◦Climate Impact
◦Sustainability and Corporate Responsibility Oversight
Corporate Responsibility for Environmental Management
We recognize that our operations have an impact on both local and global communities from the energy we source, the waste we generate, and the water we discharge. When improving and innovating our operational infrastructure across our enterprise, we give due consideration to the extent to which they may adversely threaten the environment and human health.
In accordance with our sustainability roadmap, we conducted a Climate-Related Risk Assessment to better understand potential climate-related impacts on our business under different conditions. While it did highlight additional opportunities to improve the climate resilience of our operations long-term, we believe the assessment confirmed that our current resilience measures are effective under a range of climate-related scenarios. We will continue to monitor to determine the relevant future disclosures, if any, necessary to provide transparency surrounding possible financial impacts to our company through sound governance, strategy, risk management, and performance monitoring.
AVAILABLE INFORMATION
Our common stock is traded on the New York Stock Exchange under the ticker symbol “EBS.” Our principal executive offices are located at 300 Professional Drive, Gaithersburg, Maryland 20879. Our telephone number is (240) 631-3200, and our website address is www.emergentbiosolutions.com. We make available, free of charge on our website, our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and all amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 (the "Exchange Act") as soon as reasonably practicable after we electronically file those reports with, or furnish them to, the SEC.
We also make available, free of charge on our website, the reports filed with the SEC by our executive officers, directors and 10% stockholders pursuant to Section 16 under the Exchange Act as soon as reasonably practicable after copies of those filings are provided to us by those persons. In addition, we intend to make available on our website all disclosures that are required to be posted by applicable law, the rules of the SEC or the New York Stock Exchange listing standards regarding any amendment to, or waiver of, our code of business conduct and ethics. We have included our website address as an inactive textual reference only. The information contained on, or that can be accessed through, our website is not a part of, or incorporated by reference into, this Annual Report on Form 10-K.
26
ITEM 1A. RISK FACTORS
The following risk factors and other information included in this Annual Report on Form 10-K should be carefully considered. The occurrence of any of the following risks or of unknown risks and uncertainties may adversely affect our business, operating results and financial condition.
RISK FACTOR SUMMARY
This risk factor summary contains a high-level summary of risks associated with our business. It does not contain all of the information that may be important to you, and you should read this risk factor summary together with the more detailed discussion of risks and uncertainties set forth following this summary. A summary of our risks includes, but is not limited to, the following:
•Reduced demand for and/or funding for procurement of CYFENDUS®, ACAM2000® , CNJ-016® (VIGIV), BAT® and/or BioThrax®, discontinuation of funding of our USG procurement and development contracts.
•Inability to secure follow-on product procurement contracts with the USG upon the expiration of any of our existing procurement contracts.
•Our inability to maintain quality and compliance in all of our manufacturing operations.
•Damage to, destruction of, or any unplanned disruption at our development and/or manufacturing facilities may impede our ability to manufacture our products, as well as deliver our bioservices.
•Our operations, including our use of hazardous materials, chemicals, bacteria and viruses expose us to significant potential liabilities.
•Clinical trials of product candidates are expensive and time-consuming, and their outcome is uncertain.
•We may fail to capitalize on the most scientifically, clinically or commercially promising or profitable product candidates.
•Failure to comply with complex laws and regulations pertaining to government contracts and resources required for responding to related government inquiries.
•Conditions associated with approvals and ongoing regulation of products may limit how and the extent to which we manufacture and market them.
•Failure to comply with various health care laws could result in substantial penalties.
•Failure to comply with obligations under USG pricing programs may require reimbursement for underpayments and the payment of substantial penalties, sanctions and fines.
•The extent to which we may be able to lawfully offer to sell and sell unapproved products in many jurisdictions may be unclear or ambiguous and such activities may subject us to regulatory enforcement actions.
•Development and commercialization of pharmaceutical products and our biologic products are subject to evolving competition from private and public sector competition, or biosimiliar manufacturers.
•NARCAN® (naloxone HCl) Nasal Spray and KLOXXADO® (naloxone HCI) Nasal Spray are currently subject to generic and branded competition and may be subject to additional branded and generic competition in the future.
•Biologic products may be affected by the approval and entry of follow-on biologics, or biosimilars in the United States and other jurisdictions.
•Challenges in obtaining or maintaining intellectual property rights and defense or enforcement of such rights.
•Potential discrepancies or challenges with respect to licenses, including our failure to comply with obligations under such licenses.
•Potential loss or misappropriation of proprietary information, know-how, and trade secrets which carries the risk of reducing the value of our technology and products.
•Entry of competing generic drugs upon expiration of patents and/or regulatory exclusivity or with patents no longer in force.
•The loss of sole-source suppliers or an increase in the price of inventory.
•If other parties do not perform as contractually required or as expected, we may not be able to obtain regulatory approval for or commercialize our product candidates.
27
•Unfavorable results of legal proceedings and government investigations could adversely impact our business, financial condition and results of operations.
•Our work on PHTs has exposed us to criticism and may expose us to further criticism, from the media, government personnel and others, which could further harm our reputation, negatively affect our share price, operations and our ability to attract and retain talent.
•Cybersecurity incidents involving us, our business partners, collaborators or other third parties could harm our ability to operate our business effectively in light of our heightened risk profile.
•We could face product liability exposure associated with the use of our medical products. There can be no assurance that the SAFETY Act, Public Readiness and Emergency Preparedness Act (the "PREP Act"), or other liability protections will be sufficient to limit or avoid product liability, and defending such cases requires significant resources.
•Our ability to maintain sufficient cash flow from our operations to pay our substantial debt, both now and in the future.
•Restrictions on the operation of our business and limitations on cash available for investment in our business operations as a result of our current indebtedness.
•Our ability to comply with the covenants under our Revolving Credit Facility, Term Loan Facility, Senior Unsecured Notes and any other debt agreements to which we may be a party.
•We may not be successful in identifying, structuring or acquiring businesses and products to drive our growth.
•Our failure to successfully integrate acquired businesses and/or assets into our operations and our ability to realize the benefits of such acquisitions.
•Our failure to realize the full benefits from our divestitures.
•Our business or our share price could be negatively affected as a result of the actions of stockholders.
•The price of our common stock has been and remains subject to extreme volatility.
The risk factors below contain more detailed descriptions of the risks identified above, as well as additional risks that may materially harm our business, financial condition or results of cash flows.
GOVERNMENT CONTRACTING RISKS
We currently derive, and historically derived a substantial portion of our revenue from USG procurement of CYFENDUS® , ACAM2000® , CNJ-016® (VIGIV), TEMBEXA®, BAT® and/or BioThrax®. If the USG’s demand for and/or funding for procurement of these products are substantially reduced, our business, financial condition, operating results and cash flows would be materially harmed.
We derive a substantial portion of our current and expected future revenues from USG procurement of CYFENDUS®. The success of our business and our future operating results are significantly dependent on anticipated funding for the procurement of our anthrax vaccines and the terms of such procurement by the USG, including the price per dose, the number of doses and the timing of deliveries. We have no certainty that funding will be made available for the procurement of our anthrax vaccines. If priorities for the Strategic National Stockpile (“SNS”) change generally, or as a result of the conclusion of the USG’s audit of the SNS, or with respect to the level of procurement of our anthrax vaccines, funding to procure future doses of CYFENDUS® or BioThrax® vaccines may be delayed, limited or not available, BARDA may never complete the anticipated full transition to stockpiling CYFENDUS® in support of anthrax preparedness, and our future business, financial condition, operating results and cash flows could be materially harmed.
In addition, we derive a substantial portion of our revenues from sales of ACAM2000® vaccine to the USG. In the past, the priorities of the SNS have changed with respect to ACAM2000® vaccine and if the priorities of the SNS change for ACAM® vaccine in the future or the USG decides not to exercise additional options under our ACAM2000® contract, our future business, financial condition, operating results and cash flows could be materially harmed.
As with any approved product, there is a risk that we may encounter challenges causing delays or an inability to deliver CYFENDUS®, ACAM2000®, CNJ-016® (VIGIV), TEMBEXA®, BAT® and/or BioThrax®, which may have a material effect on our ability to generate and recognize revenue.
28
Our USG procurement and development contracts require ongoing funding decisions by the USG. Any reduction or discontinuation of funding of any of these contracts could cause our business, financial condition, operating results and cash flows to suffer materially.
The USG is the principal customer for our MCMs and the primary source of funds for the development of most of our product candidates in our development pipeline. We anticipate that the USG will also be a principal customer for any MCMs that we successfully develop from within our existing product development pipeline, as well as those we acquire in the future. Additionally, a significant portion of our revenue comes from USG development contracts and grants. Over its lifetime, a USG procurement or development program, such as for CYFENDUS® under our development and procurement contract with BARDA, may be implemented through the award of many different individual contracts and subcontracts. The funding for such government programs is subject to Congressional appropriations, generally made on a fiscal year basis, even for programs designed to continue for several years. These appropriations can be subject to a number of uncertainties, including political considerations, changes in priorities due to global pandemics, the results of elections and stringent budgetary constraints.
Additionally, our government-funded development contracts typically give the USG the right, exercisable in its sole discretion, to extend these contracts for successive option periods following a base period of performance. The value of the services to be performed during these option periods may constitute the majority of the total value of the underlying contract. For example, in July 2023, we were awarded a 10-year contract by BARDA for the advanced development, manufacturing scale-up, and procurement of EbangaTM (ansuvimab-zykl) treatment for Ebola. The contract consists of a base period of performance with two option periods valued at approximately $121 million, which were exercised in September 2024 and January 2025, respectively, and option periods for procurement of EbangaTM over five years valued at up to $583 million, one of which was awarded in September 2024 (valued at $41.9 million). If all option periods are exercised, the total contract value will be valued at up to approximately $704 million. If levels of government expenditures and authorizations for public health countermeasure preparedness decrease or shift to programs in areas where we do not offer products or are not developing product candidates, or if the USG otherwise declines to exercise its options under this contract or our other existing contracts, our revenues would suffer, as well as our business, financial condition, operating results and cash flows.
There can be no assurance that we will be able to secure follow-on product procurement contracts with the USG upon the expiration of any of our existing procurement contracts.
A significant portion of our revenue is substantially dependent upon product procurement contracts with the USG and foreign governments for our MCMs and other commercialized products. Upon the expiration of a procurement contract, we may not be able to negotiate a follow-on procurement contract for the particular product on similar terms. We intend to negotiate follow-on procurement contracts for most of our MCMs and other commercialized products upon the expiration of a related procurement contract, but there can be no assurance that we will be successful obtaining any follow-on contracts. Even if we are successful in negotiating a follow-on procurement contract, it may be for a lower product volume, over a shorter period of performance or be on less favorable pricing or other terms. An inability to secure follow-on procurement contracts for our approved products or product candidates could materially and adversely affect our revenues, and our business, financial condition, operating results and cash flows could be harmed.
The government contracting process is typically a competitive bidding process and involves unique risks and requirements.
Our business involves government contracts and grants, which may be awarded through competitive bidding. Competitive bidding for government contracts presents many risks and requirements, including:
•the possibility that we may be ineligible to respond to a request for proposal;
•the commitment of substantial time and attention of management and key employees to the preparation of bids and proposals;
•the need to accurately estimate the resources and cost structure that will be required to perform any contract that we might be awarded;
•the submission by third parties of protests to contracts that we are awarded that could result in delays or withdrawals of the respective requests for proposal; and
•in the event our competitors protest or challenge contract or grant awards made to us through competitive bidding, the potential that we may incur expenses or delays, and that any such protest or challenge could result in the resubmission of bids based on modified specifications, or in the termination, reduction or modification of the awarded contract.
29
The USG may choose not to award us future contracts for either the development of our new product candidates or for the procurement of our existing MCM and other commercialized products and may instead award such contracts to our competitors. For example, ACAM2000® vaccine faces competition from JYNNEOSTM vaccine, which has been procured by the USG in recent years. If we are unable to secure particular contracts, we may not be able to operate in the market for products that are provided under those contracts. Additionally, if we are unable to consistently win new contract awards over an extended period, or if we fail to anticipate all of the costs or resources that we will be required to secure and, if applicable, perform under such contract awards, our growth strategy and our business, financial condition and operating results and cash flows could be materially and adversely affected.
The amounts we are paid under our fixed price government procurement contracts are based on estimates we have made of the time, resources and expenses required for us to perform under those contracts. If our actual costs exceed our estimates, we may not be able to earn an adequate return or may incur a loss under these contracts, which could harm our operating results and materially reduce our net income.
Our current procurement contracts with the HHS and the DoW are generally fixed price contracts. We expect that any future procurement contracts we successfully secure with the USG would likely also be fixed price contracts. Under a fixed price contract, we are required to deliver our products at a fixed price regardless of the actual costs we incur. Estimating costs that are related to performance in accordance with contract specifications is difficult, particularly where the period of performance is over several years, and when factoring in higher levels of inflation. Our failure to anticipate technical problems, estimate costs accurately or control costs during performance of a fixed price contract could reduce the profitability of such a contract or cause a loss, which could harm our operating results and materially reduce our net income.
Unfavorable provisions in government contracts, some of which may be customary, may subject our business to material limitations, restrictions and uncertainties and may have a material adverse impact on our business, financial condition, operating results and cash flows.
Government contracts customarily contain provisions that give the USG substantial rights and remedies, many of which are not typically found in commercial contracts, including provisions that allow the USG to:
•terminate existing contracts, in whole or in part, for any reason;
•unilaterally reduce or modify contracts or subcontracts;
•decline, in whole or in part, to exercise an option to purchase product under a procurement contract or to fund additional development under a development contract;
•decline to renew a procurement contract;
•claim certain rights to facilities or to products, including intellectual property, developed under the contract;
•require repayment of contract funds spent on construction of facilities in the event of contract default;
•take actions that result in a longer development timeline than expected;
•direct the course of a development program in a manner not chosen by the government contractor;
•suspend or debar the contractor from doing business with the government or a specific government agency;
•pursue civil or criminal remedies under acts such as the False Claims Act and False Statements Act; and
•control or prohibit the export of products.
Generally, government contracts contain provisions permitting unilateral termination or modification, in whole or in part, at the USG’s convenience. Under general principles of government contracting law, if the USG terminates a contract for convenience, the government contractor may recover only its incurred or committed costs, settlement expenses and profit on work completed prior to the termination. If the USG terminates a contract for default, the government contractor is entitled to recover costs incurred and associated profits on accepted items only and may be liable for excess costs incurred by the government in procuring undelivered items from another source. All of our development and procurement contracts with the USG are terminable at their convenience with these potential consequences.
In addition, our contracts with the USG grant the USG the right to use technologies developed by us under the government contract or the right to share data related to our technologies, for or on behalf of the USG. Under our contracts with the USG, we may not be able to limit third parties, including our competitors, from accessing certain of these technology or data rights, including intellectual property, in providing products and services to the USG.
30
MANUFACTURING RISKS
An inability to maintain manufacturing compliance at our manufacturing facilities, which could damage our reputation and adversely affect our business, financial condition, operating results and cash flows.
The FDA conducts periodic inspections of our manufacturing facilities for compliance with CGMP requirements. The Company's failure to maintain compliance with CGMP requirements at our manufacturing facilities has hindered and could continue to hinder our ability to continue manufacturing for our own products and for Bioservices customers, which could adversely affect our business, financial condition, operating results and cash flows.
The failure to remedy any objectionable conditions at Emergent's manufacturing facilities, any additional failures to maintain compliance with CGMP requirements at any of our manufacturing facilities, or any administrative or regulatory action or recommendation to take any such action by the FDA could damage our reputation and adversely affect our business, financial condition, operating results and cash flows.
Damage to, destruction of, or any disruption at our manufacturing facilities could impede our ability to manufacture our products or product candidates, as well as impact the delivery of bioservices to third parties, which would harm our business, financial condition, operating results and cash flows.
Any interruptions in our manufacturing operations could result in our inability to produce products and product candidates for delivery to satisfy the demands of our customers in a timely manner, which would reduce our revenues and materially harm our business, financial condition, operating results and cash flows. A number of factors could cause interruptions, including:
•equipment malfunctions or failures;
•technology malfunctions;
•cyber-attacks;
•work stoppages or slowdowns;
•civil unrest and protests, including by animal rights activists;
•litigation, investigations or government enforcement actions;
•damage to or destruction of our manufacturing equipment, or of one or more of our facilities;
•findings and recommendations of health authorities or qualified persons in connection with facility inspections;
•ongoing supply chain interruptions; and
•product contamination or tampering.
The factors listed above could cause disruptions at any of our manufacturing facilities. We do not have any redundant manufacturing facilities for any of our products. Accordingly, any damage to, or destruction of, or disruption at one or more of our facilities could impede our ability to manufacture our products, and our product candidates and our ability to provide manufacturing and development services for external customers, result in losses and delays, including delays in the performance of our contractual obligations or delays in our clinical trials, any of which could be costly to us and materially harm our business, financial condition, operating results and cash flows.
Providers of MCMs could be subject to an increased risk of terrorist activities. The USG has designated our Lansing, Michigan facility as requiring additional security. Although we continually evaluate and update security measures, there can be no assurance that any additional security measures would protect these facilities from terrorist efforts determined to disrupt our manufacturing activities.
31
Problems may arise during the production of our products and product candidates, as well as those we produce for our Bioservices customers, due to the complexity of the processes involved in product development, manufacturing and shipment or other factors. Significant delays in product development or manufacturing and our ability to ramp up production to meet the needs of our customers could cause delays in recognizing revenues, which would harm our business, financial condition, operating results and cash flows.
The majority of our products and product candidates are biologics. Manufacturing biologics, especially in large quantities, is complex. The products must be made consistently and in compliance with a clearly-defined manufacturing process. Problems during manufacturing may arise for a variety of reasons, including problems with raw materials, equipment malfunction and failure to follow specific protocols and procedures. Slight deviations anywhere in the manufacturing process, including obtaining materials, maintaining master seed or cell banks and preventing genetic drift, seed or cell growth, fermentation, contamination including from particulates among other things, filtration, filling, labeling, packaging, storage and shipping, potency and stability issues and other quality control testing, may result in lot failures or manufacturing shut-downs, delays in the release of lots, product recalls, spoilage or regulatory action. Such deviations may require us to alter manufacturing processes or change manufacturers. Additionally, as our equipment ages, it will need to be replaced, which has the potential to result in similar consequences. Success rates can also vary dramatically at different stages of the manufacturing process, which can reduce yields and increase costs. From time to time, we may experience deviations in the manufacturing process, including as a result of regulatory action, that may take significant time and resources to resolve and, if unresolved, may affect manufacturing output and could cause us to fail to satisfy customer orders or contractual commitments, lead to a termination of one or more of our contracts, lead to delays in our clinical trials, result in litigation, or result in other restrictions on the marketing or manufacturing of a product, any of which could be costly to us, damage our reputation and negatively impact our business.
Additionally, if changes are made to the manufacturing process, we may be required to provide the FDA with pre-clinical and clinical data showing the comparable identity, strength, quality, purity or potency of any impacted products before and after the changes.
We are contractually required to ship our biologic products at a prescribed temperature range and variations from that temperature range could result in loss of product and could significantly and adversely impact our revenues, which would harm our business, financial condition, operating results and cash flows.
In addition, we may not be able to ramp up our manufacturing processes to meet the rapidly changing demand or specifications of our customers on the desired timeframe, if at all. Our inability to ramp up manufacturing to meet the demand or specifications of our customers or the inability to timely obtain regulatory authorization to produce the products or product candidates of our customers could also harm our business, financial condition, operating results and cash flows.
Our products and product candidates procured by the USG and other customers require us to perform tests for and meet certain product release standards prescribed by the FDA and other agencies, which may not be met on a timely basis or at all.
We are unable to sell any products and product candidates that fail to satisfy certain testing specifications. For example, we must provide the FDA with the results of certain tests, including potency tests, before certain lots are released for sale. Potency testing of each applicable lot is performed against qualified control lots that we maintain. We continually monitor the status of such reference lots for FDA compliance and periodically produce and qualify a new reference lot to replace the existing reference lot. If we are unable to satisfy regulatory authority and/or USG requirements for the release of our products or product candidates, our ability to supply such products and product candidates to authorized buyers would be impaired until such time as we become able to meet such requirements, which could materially harm our future business, financial condition, operating results and cash flows.
32
Our operations, including our use of hazardous materials, chemicals, bacteria and viruses, require us to comply with regulatory requirements and expose us to significant potential liabilities.
Our operations involve the use of hazardous materials, including chemicals, bacteria and viruses, and may produce dangerous waste products. Accordingly, we, along with the third parties that conduct clinical trials and manufacture our products and product candidates on our behalf, are subject to federal, state, local and foreign laws and regulations that govern the use, manufacture, distribution, storage, handling, exposure, disposal and recordkeeping with respect to these materials. Under the Federal Select Agent Program, pursuant to the Public Health Security and Bioterrorism Preparedness and Response Act, we are required to register with and be inspected by the Centers for Disease Control and Prevention (the "CDC") and the Animal and Plant Health Inspection Service if we have in our possession, or if we use or transfer, select biological agents or toxins that could pose a threat to public health and safety, to animal or plant health or to animal or plant products. This legislation requires stringent safeguards and security measures for these select agents and toxins, including controlled access and the screening of entities and personnel and establishes a comprehensive national database of registered entities. We are also subject to a variety of environmental and occupational health and safety laws. Compliance with current or future laws and regulations in this area can require significant costs and we could be subject to substantial fines and penalties in the event of noncompliance. In addition, the risk of contamination or injury from these materials cannot be completely eliminated. In such event, we could be held liable for substantial civil damages or costs associated with the cleanup of hazardous materials. From time to time, we have been involved in remediation activities and may be so involved in the future. Any related cost or liability might not be fully covered by insurance, could exceed our resources and could have a material adverse effect on our business, financial condition, operating results and cash flows. In addition to complying with environmental and occupational health and safety laws, we must comply with special regulations relating to biosafety administered by the CDC, HHS, U.S. Department of Agriculture and the DoW, as well as regulatory authorities in Canada.
PRODUCT DEVELOPMENT AND COMMERCIALIZATION RISKS
The product candidates that we work on internally or for third-party customers may not be safe or effective and even if they are, we may be unable to manufacture sufficient quantities to meet demand.
We provide bioservices for the development and/or manufacture of various product candidates. There can be no assurance that these product candidates will be safe or effective or that they will be authorized for emergency use or approved by the FDA or any other health regulatory authority. Even if product candidates are found to be safe and/or effective and receive authorization or approval by a health regulatory authority or we receive authorization to produce drug substance or drug product at our facilities, the manufacturing processes for our Bioservices programs are complex. There can be no assurance that we will be able to produce sufficient clinical or commercial quantities of any product candidate in a timely basis or at all. Further, as we have reduced the emphasis on our Bioservices business, concerns may exist regarding our ability to fulfill manufacturing commitments to our Bioservices customers. Any future failure to satisfy manufacturing commitments could adversely affect our reputation, subject us to potential legal liability and harm our business, financial condition, operating results and cash flows.
Our growth depends on our success in developing and commercializing our product candidates. If we are unable to commercialize these product candidates or experience significant delays or unanticipated costs in doing so, our business would be materially and adversely affected.
We have invested significant efforts and financial resources in the development of our vaccines, therapeutics and medical device product candidates and the acquisition of additional product candidates. In addition to our product sales, our ability to generate revenue is dependent on a number of factors, including the success of our development programs, the USG's interest in providing development funding for or procuring certain of our product candidates, and the commercial viability of our acquired or developed product candidates. The commercial success of our product candidates can depend on many factors, including accomplishing the following in an economical manner:
•successful development, formulation and CGMP or Quality Management System Regulation ("QMSR") scale-up of manufacturing that meets FDA and/or foreign regulatory requirements;
•successful program partnering;
•successful completion of clinical or non-clinical development;
•receipt of marketing approvals, clearances, or other authorizations from the FDA and equivalent foreign regulatory authorities;
•establishment of commercial manufacturing processes and product supply arrangements;
•training of a commercial sales force for the product;
33
•successful registration and maintenance of relevant patent and/or other proprietary protection;
•competitive pricing and market access; and
•acceptance of the product by potential government and other customers.
In particular, the success of NARCAN® (naloxone HCl) Nasal Spray, including in over-the-counter form, is subject to commercial availability of the product and our ability to gain sufficient market acceptance by physicians, patients, third-party payors and others in the medical community.
Clinical trials of product candidates are expensive and time-consuming, and their outcome is uncertain. We must invest substantial amounts of time and financial resources in these trials, which may not yield viable products. Failure to obtain regulatory approval for product candidates, particularly in the United States, could materially and adversely affect our financial resources, which would adversely affect our business, financial condition, operating results and cash flows.
Before obtaining regulatory approval or other authorization of our product candidates, we and our collaborative partners, where applicable, must conduct pre-clinical studies and clinical trials to establish proof of concept and demonstrate the safety and efficacy of our product candidates. Pre-clinical and clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. Success in pre-clinical testing and early clinical trials does not ensure that later clinical trials will be successful, and interim results of such trials do not necessarily predict final results. An unexpected result in one or more of our clinical trials can occur at any stage of testing.
We may experience unforeseen events or issues during, or as a result of, pre-clinical testing, clinical trials or animal efficacy studies. These issues and events, which could delay or prevent our ability to receive regulatory approval for a product candidate, include, among others:
•our inability to manufacture sufficient quantities for use in trials;
•the unavailability or variability in the number and types of subjects for each study;
•safety issues or inconclusive or incomplete testing, trial or study results;
•drug immunogenicity;
•lack of efficacy of product candidates during trials;
•government or regulatory restrictions or delays; and
•greater than anticipated costs of trials.
Pre-clinical and clinical testing for certain of our MCM product candidates may face additional difficulties and uncertainties because they cannot ethically or feasibly be tested in human subjects. In the U.S. we expect to rely on the Animal Rule to obtain regulatory approval for some of our MCM product candidates. The Animal Rule permits, for certain limited diseases and circumstances, the use of animal efficacy studies, together with human clinical safety and immunogenicity trials, to support an application for marketing approval. For a product approved under the Animal Rule, certain additional post-marketing requirements apply. For example, to the extent feasible and ethical, applicants must conduct post-marketing clinical studies, such as field studies in the event of an outbreak or act of bioterrorism, to assess the drug's safety and effectiveness. It is possible that results from the animal efficacy studies used to support approval under the Animal Rule may not be predictive of the actual efficacy of our product candidates in humans.
Under the Public Health Service Act (the "PHSA") and the Federal Food, Drug, and Cosmetic Act (the "FDCA"), the Secretary of HHS can contract to purchase MCMs for the SNS prior to FDA approval, clearance, or other authorization of certain MCM product candidates. If the USG does not provide funding for and procure our MCM product candidates, they generally will have to be approved by the FDA through traditional regulatory mechanisms prior to sale and distribution in the United States.
We may fail to select or capitalize on the most scientifically, clinically or commercially promising or profitable product candidates.
We continue to evaluate our product development strategy and, as a result, may modify our strategy in the future. In this regard, we may, from time to time, focus our product development efforts on different product candidates or may delay or halt the development of various product candidates. As part of our stabilization efforts in 2024 related to our multi-year strategic plan, we changed and refocused several areas of our business, and may continue to change or refocus our business activities, including product development, commercialization and manufacturing activities based on government funding decisions and other factors. This required changes, and may require changes in the future, in our facilities and our personnel.
34
Any product development changes that we implement may not be successful. In particular, we may fail to select or capitalize on the most scientifically, clinically or commercially promising or profitable product candidates or we may choose candidates for which government development funds are not available. Our decisions to allocate our R&D, management and financial resources toward particular product candidates or therapeutic areas may not lead to the development of viable commercial products and may divert resources from better business opportunities. Similarly, our decisions to delay or terminate product development programs could also cause us to miss valuable opportunities.
REGULATORY AND COMPLIANCE RISKS
There are a number of complex laws and regulations that pertain to government contracts and compliance with those laws and regulations require significant time and cost, which could have a material adverse effect on our business, financial condition, operating results and cash flows.
As a manufacturer and supplier of MCMs and other approved products to the USG addressing PHTs, we must comply with numerous laws and regulations relating to the procurement, formation, administration and performance of government contracts. These laws and regulations govern how we operate our business and transact business with our government clients and impose compliance costs on our operations. In addition, these laws and regulations may be subject to sudden change, which may impose additional costs and related obligations on our operations or require us to make changes to the way we operate our business or transact business with our government clients. For a detailed description of the most significant regulations that affect our government contracting business, see the discussion under “Regulation - Government Contracting” elsewhere in this Annual Report.
We may be subject to government investigations into compliance with government acquisition regulations. USG agencies routinely audit and investigate government contractors for compliance with applicable laws and standards. Even though we take significant precautions to identify, prevent and deter fraud, misconduct and non-compliance, we face the risk that our personnel or outside partners may engage in misconduct, fraud or improper activities. If we are audited or investigated and such audit or investigation were to uncover improper or illegal activities, we could be subject to civil and criminal fines and penalties, and administrative sanctions, including suspension or debarment from government contracting, and we could suffer significant reputational harm. The loss of our status as an eligible government contractor or significant fines or penalties associated with contract non-compliance or resulting from investigations could have a material adverse effect on our business.
Our long-term success depends, in part, upon our ability to develop, receive regulatory approval for and commercialize product candidates we develop or acquire and, if we are not successful, our business, financial condition, operating results and cash flows may suffer.
Our product candidates and the activities associated with them in the U.S. are subject to extensive FDA regulation and oversight. This includes, but is not limited to, laws and regulations governing product development, product labeling, product testing, manufacturing, storage, product distribution, record keeping, and advertising and promotion. In limited circumstances, governments may have the authority to procure products that have not obtained regulatory approval to stockpile for emergency preparedness and to respond to public health emergencies. In other circumstances, failure to obtain regulatory approval for a product candidate will prevent us from selling and commercializing the product candidate.
35
In the United States, to obtain authorization from the FDA to market and sell any of our future drug, biologic, or vaccine products, we will be required to submit an NDA or BLA to the FDA. Under the FDCA, the PHSA, and the FDA’s implementation of those statutes, a company must support an NDA or BLA with substantial evidence that the product candidate is effective and evidence that the product is safe. Ordinarily, the FDA requires data from adequate and well-controlled clinical trials, including Phase 3 trials conducted in patients with the disease or condition being targeted, to demonstrate that a drug meets the statutory standards for approval. Once an NDA or BLA is submitted, the FDA has substantial discretion and may refuse to accept our application or may decide that our data are insufficient to support approval and require additional pre-clinical, clinical or other studies. Even if marketing approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed, or to conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. Likewise, changes to our combination products, including changes to the device constituent part, may also require a new submission to, and approval from, the FDA. The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory and policy changes. Average review times at the FDA have fluctuated in recent years as a result. Disruptions at the FDA and other agencies may also slow the time necessary for new product candidates to be reviewed and approved by necessary government agencies, which could adversely affect our business. In addition, government funding of other government agencies on which our operations may rely, including those that fund research and development activities, is subject to the political process, which is inherently fluid and unpredictable. Additional delays may result if an FDA Advisory Committee or other regulatory authority recommends non-approval or restrictions on approval. In addition, we may experience delays or rejections based on additional government regulation from future legislation or administrative action or based on changes in regulatory authority policy during the period of product development, clinical trials and the review process.
Our MCM product candidates may be eligible for approval under the FDA's “Animal Rule,” under which findings from adequate and well controlled animal efficacy studies may serve as the basis of an approval when it is not feasible or ethical to conduct efficacy trials in humans. We cannot guarantee that the FDA will permit us to proceed with approval or licensure of any of our MCM product candidates under the Animal Rule. Even if we are able to proceed under the Animal Rule, product development can take a considerable amount of time, and the FDA may decide that our data are insufficient to support approval and require additional pre-clinical, clinical or other studies, refuse to approve our products, or place restrictions on our ability to commercialize those products. Furthermore, products approved under the Animal Rule are subject to certain additional post-marketing requirements. We cannot guarantee that we will be able to meet this regulatory requirement even if one or more of our product candidates are approved under the Animal Rule.
The process of obtaining these regulatory approvals is expensive, often takes many years if approval is obtained at all, and can vary substantially based upon the type, complexity and novelty of the product candidate involved. Changes in the regulatory approval process may cause delays in the approval or other marketing authorization, or rejection of an application. There is a high rate of failure inherent in the medical product development process, and product candidates that appear promising at early stages of development may fail for a number of reasons. Moreover, positive results from pre-clinical studies may not be predictive of similar results in human clinical trials and promising results from earlier clinical trials of a product candidate may not be replicated in later clinical trials.
Failure to successfully develop future product candidates may materially adversely affect our business, financial condition, operating results and cash flows.
Unapproved and investigational stage products are also subject to the FDA's laws and regulations governing advertising and promotion, which prohibit the promotion of both unapproved products and unapproved uses of approved products. There is some risk that the FDA could conclude that our communications relating to unapproved products or unapproved uses of approved products constitute the promotion of an unapproved product or product use in violation of FDA laws and regulations. There is also a risk that a regulatory authority in another country could take a similar position under that country's laws and regulations and conclude that we have violated the laws and regulations related to product development, approval, or promotion in that country. If the FDA or any foreign regulatory authority determines that any of our communications constitute pre-approval promotion or promotion of an off-label use, the FDA could request that we modify our promotional materials, issue an untitled letter or warning letter, or subject us to regulatory or enforcement actions, including injunction, seizure, civil fine or criminal penalties.
36
Even if we or our collaborators obtain marketing approvals for our product candidates, the conditions of approvals and ongoing regulation of our products may limit how we manufacture, market and sell our products, which could materially impair our ability to generate revenue.
Once marketing authorization has been granted, we and our business partners will remain subject to ongoing regulatory oversight of our medical products, including with respect to labeling; safety surveillance and reporting; registration and listing requirements; CGMP and QMSR requirements relating to manufacturing, quality control, quality assurance, and corresponding maintenance of records and documents; advertising and promotional activities; requirements regarding the distribution of samples to physicians and related recordkeeping; and medical device design, development and manufacturing. In February 2024, FDA issued the Quality Management System Regulation (QMSR) Final Rule to amend the QSR, incorporating by reference ISO 13485:2016. The QMSR became effective on February 2, 2026.
The FDA and other agencies, including the U.S. Department of Justice (“DOJ”) and the HHS Office of Inspector General (“OIG”), closely regulate and monitor the marketing and promotion of medical products to ensure that they are marketed in a manner consistent with the FDA-approved label and regulatory authorities in other countries may have similar policies. For drug products, we must promote the product in a manner consistent with the full prescribing information or, for devices, consistent with the approved or cleared indications for use. The FDA, DOJ, and OIG impose stringent restrictions on manufacturers’ communications regarding unapproved/uncleared products and unapproved/uncleared uses of approved/cleared products. If we market unapproved/uncleared products or market our approved/cleared products for unapproved/uncleared indications, we may be subject to enforcement action.in the U.S. or foreign countries, including civil and administrative remedies (such as entering into corporate integrity agreements with the USG), as well as criminal sanctions. If our employees or agents engage in marketing of an unapproved/uncleared product or the unapproved/uncleared use of an approved/cleared product, we could be subject to civil or criminal investigations and monetary and injunctive penalties, which could adversely impact our ability to conduct business in certain markets, negatively affect our business, financial condition, operating results and cash flows, and damage our reputation.
Violations of the FDCA and other statutes, including the False Claims Act, relating to the promotion and advertising of prescription products may lead to investigations and enforcement actions alleging violations of federal and state health care fraud and abuse laws, as well as state consumer protection laws. Moreover, if the FDA or any foreign regulatory authority determines that any of our communications constitute pre-approval promotion or promotion of an off-label use, the FDA or foreign regulatory authority could request that we modify our promotional materials, issue an untitled letter or warning letter or the foreign equivalent, or subject us to regulatory or enforcement actions, including injunction, seizure, civil fine or criminal penalties.
Certain of our products are subject to post-marketing requirements (“PMRs”), which we are required to conduct, and post marketing commitments, which we have agreed to conduct. The FDA has the authority to take action against sponsors who fail to meet the obligations of a PMR, including civil monetary penalties and/or misbranding charges.
In addition, discovery of previously unknown adverse events or other problems with our products, manufacturing partners or manufacturing processes, or failure to comply with regulatory requirements, may result in various penalties and sanctions. For all FDA-regulated products, if the FDA finds that a manufacturer has failed to comply with applicable laws and regulations, or that a product is ineffective or poses an unreasonable health risk, it can institute or seek a wide variety of enforcement actions and other remedies, including but not limited to:
•restrictions on such products, manufacturers or manufacturing processes;
•restrictions on the labeling or marketing of a product;
•restrictions on distribution or use of a product;
•requirements to conduct post-marketing studies or clinical trials;
•warning letters or untitled letters;
•refusal to approve pending applications or supplements to approved applications that are submitted;
•delay in or refusal to approve, clear or authorize pending PMA applications, 510(k) premarket submissions, or de novo classification requests;
•fines, restitution or disgorgement of profits or revenues;
•suspension or withdrawal of marketing approvals;
•refusal to permit the import or export of our products;
37
•product seizure; and
•injunctions or the imposition of civil or criminal penalties.
If we and our collaborators are not able to comply with post-approval regulatory requirements, we could have the marketing approvals for our products withdrawn by regulatory authorities and our ability to market and sell any products could be limited, which could adversely affect our ability to achieve or sustain profitability. Further, the cost of compliance with post-approval regulations may have a negative effect on our operating results and financial condition.
Any product candidate for which we or our collaborators obtain marketing approval could be subject to restrictions or withdrawal from the market and we may be subject to substantial penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our product candidates, when and if any of them are approved.
Likewise, non-compliance with EU, any EU Member State, or UK requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, can also result in significant financial penalties. Similarly, failure to comply with the European and other legal and regulatory requirements regarding the protection of personal information can also lead to significant penalties and sanctions. Non-compliance with similar requirements in other foreign jurisdictions can also result in enforcement actions and significant penalties.
Current and future policy or legislation may increase the difficulty and cost for us and our collaborators to obtain marketing approval of and commercialize our product candidates, which may affect the prices we, or our collaborators, may obtain.
In the United States and foreign jurisdictions, there have been a number of policy, legislative and regulatory changes and proposed changes regarding the health care system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any approved products. We expect that current laws, as well as other health care reform measures that may be adopted in the future, may result in more rigorous coverage criteria and additional downward pressure on the price that we, or any collaborators, may receive for any approved products.
There has been continued heightened federal governmental scrutiny over the manner in which manufacturers set prices for their marketed products. This includes Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products.
Further, the Inflation Reduction Act of 2022 (the “IRA”) requires manufacturers of certain Part B and Part D drugs to issue to HHS rebates based on certain calculations and triggers (i.e., when drug prices increase and outpace the rate of inflation). While we are not directly affected by the IRA at this time, these types of laws may have a significant impact on our ability to set a product price we believe is fair and may adversely affect our ability to generate revenue and achieve or maintain profitability.
A number of states have passed legislation and implemented regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. A number of states, for example, require drug manufacturers and other entities in the drug supply chain, including health carriers, pharmacy benefit managers, and wholesale distributors, to disclose information about pricing of pharmaceuticals. These measures could reduce the ultimate demand for approved products, or put pressure on our product pricing. We expect that additional state and federal health care reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for health care products and services, which could result in reduced demand for our products or additional pricing pressures.
38
If we fail to comply with foreign, federal, state and local health care laws, including fraud and abuse laws, health information privacy and security laws, and antitrust laws, we could face substantial penalties and our business, results of operations, financial condition and prospects could be adversely affected.
In the United States, certain of our products are reimbursed under federal and state health care programs such as Medicaid, Medicare, Tricare, and/or state pharmaceutical assistance programs. Many foreign countries have similar laws. Federal and state laws designed to prevent fraud and abuse under these programs prohibit pharmaceutical companies from offering valuable items or services to customers or potential customers to induce them to buy, prescribe, or recommend our product (the so-called “anti-kickback” laws). Exceptions are provided for discounts and certain other arrangements if specified requirements are met. Other federal and state laws, and similar foreign laws, not only prohibit us from submitting any false information to government reimbursement programs but also prohibit us, our employees, or any third party acting on our behalf from doing anything to cause, assist, or encourage our customers to submit false claims for payment to these programs. We are also subject to various federal, state and foreign antitrust and competition laws that prohibit certain activities that may have an impact against potential competitors. Violations of the various fraud and abuse and antitrust laws may result in severe penalties against the responsible employees and us, including jail sentences, large fines, and the exclusion of our products from reimbursement under federal and state programs. Some of the laws that may affect our ability to operate include:
•the federal Anti-Kickback Statute makes it illegal for any person or entity, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, receive, offer or pay remuneration, directly or indirectly, overtly or covertly, to induce, or in return for, either the referral of an individual, or the purchase, lease, prescribing or recommendation of an item, good, facility or service reimbursable by a federally funded health care program, such as the Medicare or Medicaid program. The term “remuneration” has been interpreted broadly and may constrain our marketing practices, educational programs, pricing policies and relationships with health care providers or other entities, among other activities;
•the federal False Claims Act imposes criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for, among other things, knowingly presenting, or causing to be presented, false or fraudulent claims for payment by a federal health care program or making a false statement or record material to payment of a false claim or avoiding, decreasing or concealing an obligation to pay money to the federal government, with potential liability, including mandatory treble damages and significant per-claim penalties.
•the U.S. federal Health Insurance Portability and Accountability Act of 1996 ("HIPAA"), which imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any health care benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any health care benefit program, regardless of the payor (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statement, in connection with the delivery of, or payment for, health care benefits, items or services. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
•HIPAA, as amended by HITECH, and their respective implementing regulations mandates, among other things, the adoption of uniform standards for the electronic exchange of information in common health care transactions, as well as standards relating to the privacy, security and transmission of individually identifiable health information, which require the adoption of administrative, physical and technical safeguards to protect such information. Among other things, HITECH makes HIPAA's security standards directly applicable to “business associates,” or independent contractors or agents of covered entities that create, receive or obtain protected health information in connection with providing a service for or on behalf of a covered entity;
•the Physician Payments Sunshine Act and its implementing regulations require certain manufacturers of drugs, biologics, medical devices and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program to report certain payments and transfers of value made to U.S. physicians, other prescribers and teaching hospitals, as well as ownership or investment interests held by physicians, and their immediate family members; and
39
•state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third-party payor, including commercial insurers; state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; state, local and foreign laws that require pharmaceutical companies to comply with the pharmaceutical industry’s otherwise voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, obtain pharmaceutical agent licensure, and/or otherwise restrict payments that may be made to health care providers and entities; and state, local and foreign laws that require drug manufacturers to report information related to payments and other transfers of value to health care providers or entities, or marketing expenditures.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available under the federal Anti-Kickback Statute, it is possible that some of our business activities could be subject to challenges under one or more of such laws.
If our operations are found to be in violation of any of the laws described above or otherwise, we may be subject to penalties, including civil and criminal penalties, damages, fines, individual imprisonment, integrity obligations, exclusion from federal health care programs and the curtailment or restructuring of our operations. Any such penalties could adversely affect our financial results. We continue to improve our corporate compliance program designed to ensure that our development, marketing, and sales of existing and future products and product candidates are in compliance with all applicable laws and regulations, but we cannot guarantee that this program will protect us from governmental investigations or other actions or lawsuits stemming from a failure to comply with such laws or regulations. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Efforts to ensure that our business arrangements with third parties comply with healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving fraud and abuse or other health care laws and regulations. If our operations are found to be in violation of any of these laws, we may be subject to significant civil, criminal and administrative penalties, damages, fines, individual imprisonment, integrity obligations, exclusion from government funded health care programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If a third party fails to comply with applicable laws and regulations while acting on our behalf, we may also be subject to criminal, civil, and administrative penalties, including those listed above.
The United States government, state governments and private payors regularly investigate the pricing and competitive practices of pharmaceutical companies and biotechnology companies, and many file actions alleging that inaccurate reporting of prices has improperly inflated reimbursement rates. We may also be subject to investigations related to our pricing practices. Regardless of merit or eventual outcome, these types of investigations and related litigation can result in:
•diversion of management time and attention;
•significant legal fees and payment of damages or penalties;
•limitations on our ability to continue certain operations;
•decreased product demand; and
•injury to our reputation.
Moreover, an adverse outcome, or the imposition of penalties or sanctions for failing to comply with applicable fraud and abuse and antitrust laws, could adversely affect us and may have a material adverse effect on our business, results of operations, financial condition and cash flows.
If we fail to comply with our obligations under U.S. governmental pricing programs, we could be required to reimburse government programs for underpayments and could pay penalties, sanctions and fines.
The issuance of regulations and coverage expansion by various governmental agencies relating to the Medicaid rebate program will continue to increase our costs and the complexity of compliance and will be time-consuming. Because we participate in the Medicaid rebate program, we are also required to report average sales price ("ASP"), information to CMS for certain categories of drugs that are paid for under Part B of the Medicare program. Future statutory or regulatory changes or CMS binding guidance could affect the ASP calculations for our products and the resulting Medicare payment rate and could negatively impact our results of operations.
40
Pricing and rebate calculations vary among products and programs, involve complex calculations and are often subject to interpretation by us, governmental or regulatory agencies and the courts. The Medicaid rebate amount is computed each quarter based on our submission to CMS of our current average manufacturer price (“AMP”) and “best price” for the quarter. If we become aware that our reporting for a prior quarter was incorrect, or has changed as a result of recalculation of the pricing data, we are obligated to resubmit the corrected data for a period not to exceed twelve quarters from the quarter in which the data originally were due. Any such revisions could have the impact of increasing or decreasing our rebate liability for prior quarters, depending on the direction of the revision. Such restatements and recalculations would increase our costs for complying with the laws and regulations governing the Medicaid rebate program. Price recalculations also may affect the “ceiling price” at which we are required to offer our products to certain covered entities, such as safety-net providers, under the 340B/Public Health Service ("PHS") drug pricing program.
In addition, if we are found to have made a misrepresentation in the reporting of ASP, we are subject to civil monetary penalties for each such price misrepresentation and for each day in which such price misrepresentation was applied. If we are found to have knowingly submitted false AMP or “best price” information to the government, we may be liable for civil monetary penalties per item of false information. Any refusal of a request for information or knowing provision of false information in connection with an AMP survey verification would also subject us to civil monetary penalties. In addition, our failure to submit monthly/quarterly AMP or “best price” information on a timely basis could result in a civil monetary penalty per day for each day the information is late beyond the due date. Such failure could also be grounds for CMS to terminate our Medicaid drug rebate agreement, under which we participate in the Medicaid program. In the event that CMS terminates our rebate agreement, no federal payments would be available under Medicaid or Medicare Part B for our covered outpatient drugs. Governmental agencies may also make changes in program interpretations, requirements or conditions of participation, some of which may have implications for amounts previously estimated or paid. We cannot ensure that CMS will not find our submissions to be incomplete or incorrect.
In order for our products to be reimbursed by the primary federal governmental programs, we must report certain pricing data to the USG. Compliance with reporting and other requirements of these federal programs is a pre-condition to: (i) the availability of federal funds to pay for our products under Medicaid and Medicare Part B; and (ii) procurement of our products by the Department of Veterans Affairs ("DVA"), and by covered entities under the 340B/PHS program. The pricing data reported are used as the basis for establishing Federal Supply Schedule ("FSS"), and 340B/PHS program contract pricing and payment and rebate rates under the Medicare Part B and Medicaid programs, respectively. Pharmaceutical companies have been prosecuted under federal and state false claims laws for submitting inaccurate and/or incomplete pricing information to the government that resulted in increased payments made by these programs. Although we maintain and follow strict procedures to ensure the maximum possible integrity for our federal pricing calculations, the process for making the required calculations is complex, involves some subjective judgments and the risk of errors always exists, which creates the potential for exposure under the false claims laws. If we become subject to investigations or other inquiries concerning our compliance with price reporting laws and regulations, and our methodologies for calculating federal prices are found to include flaws or to have been incorrectly applied, we could be required to pay or be subject to additional reimbursements, penalties, sanctions or fines, which could have a material adverse effect on our business, financial condition and results of operations.
To be eligible to have our products paid for or reimbursed with federal funds under the Medicaid and Medicare Part B programs and purchased by certain federal agencies and grantees, we also must participate in the DVA FSS pricing program. To participate, we are required to enter into an FSS contract with the DVA, under which we must make our innovator “covered drugs” available to all federal purchasers. In addition, for the “Big Four” federal agencies—the DVA, the DoW, the PHS (including the Indian Health Service), and the Coast Guard—we must make covered drugs available at pricing that is capped at the statutory federal ceiling price ("FCP"). The FCP is calculated using the formula set forth in Section 603 of the Veterans Health Care Act of 1992 (the "VHCA") and based on a weighted average wholesale price known as the Non-Federal Average Manufacturer Price ("Non-FAMP"), which manufacturers are required to report on a quarterly and annual basis to the DVA. Under the VHCA, knowingly providing false information in connection with a Non-FAMP filing can subject us to significant penalties for each item of false information. If we overcharge the government in connection with our FSS contract or Tricare program agreements, whether due to a misstated FCP or otherwise, we are required to disclose the error and refund the difference to the government. The failure to make necessary disclosures and/or to identify contract overcharges can result in allegations against us under the False Claims Act and other laws and regulations. Unexpected refunds to the government, and responding to a government investigation or enforcement action, can be expensive and time-consuming, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
41
From time to time, we sell unapproved MCMs to government entities under certain circumstances. While this is permissible in some cases, the extent to which we may be able to lawfully offer to sell and sell unapproved products in many jurisdictions may be unclear or ambiguous. Such sales could subject us to regulatory enforcement action, product liability and reputational risk.
Under certain and narrow circumstances, MCMs may be procured by government entities prior to approval by the FDA or other U.S. regulatory authorities, a practice which we followed in connection with CYFENDUS® prior to its approval by the FDA. In the United States, the Secretary of HHS has the authority to contract to purchase MCMs for the SNS prior to FDA approval of the relevant MCM in specified circumstances. The FDA also has the authority to permit the emergency use of medical products that have not yet been approved by the FDA under an EUA. An EUA terminates when the EUA is revoked or the emergency declaration underlying the EUA terminates. However, an EUA is not a long-term alternative to obtaining FDA approval, licensure, clearance, or other marketing authorization for a product. Absent an applicable exception, our MCM product candidates generally will have to be approved, licensed, or cleared by the FDA or other regulatory authorities in the relevant country through traditional pathways before we can sell those products to governments. Additionally, the laws in certain jurisdictions regarding the ability of government entities to purchase unapproved product candidates can be ambiguous, and the permissibility of exporting unapproved products from the United States and importing them to foreign countries may be unclear in some instances. Nevertheless, government bodies, such as U.S. federal entities other than HHS, state and local governments within the United States, and foreign governments have sought and may further seek to procure our MCM product candidates that are not yet approved. In this situation, we would expect to assess the permissibility and liability implications of supplying our product candidates to such entities on a case-by-case basis, which presents certain challenges, both in the case of U.S. and foreign governments, and particularly under emergency conditions. In addition, agencies or branches of one country’s government may take different positions regarding the permissibility of such sales than another country’s government or even other agencies or branches of the same government. If local enforcement authorities disagree with our conclusion that such activities are permissible, they may take enforcement action against us.
In addition, the sale of unapproved products also could give rise to product liability claims for which we may not be able to obtain adequate indemnification or insurance coverage. For example, despite liability protections applicable to claims arising under U.S. law and resulting from the use of certain unlicensed or unauthorized MCMs, such as a declaration issued under the PREP Act, plaintiffs still may bring lawsuits alleging, among other things, that their claims are not barred under the PREP Act.
In the event that a user of one or more of our products experiences an adverse event, we may be subject to additional reputational risk if the product has not been approved by the FDA or the corresponding regulatory authority of another country, particularly because we will not have approved labeling regarding the safety or efficacy of those products. In addition, legislatures and other governmental bodies that have oversight responsibility for procuring agencies may raise concerns after the fact, even if procurement was permissible at the time, which could result in negative publicity, reputational risk and harm to our business prospects.
There is also a risk that our communications with governments about our unapproved/uncleared products, such as in the procurement context, could be considered promotion of an unapproved/uncleared product or unapproved/uncleared use of an approved product. Therefore, there is a risk that we could be subject to enforcement actions if found to be in violation of such laws or regulations.
Even after regulatory approval is received, if we fail to comply with regulatory requirements, or if we experience unanticipated problems with our approved products, they could be subject to restrictions, penalties or withdrawal from the market.
Any vaccine, therapeutic product or medical device for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for such product, will be subject to the continual requirements of and review by the FDA and other regulatory bodies. Our approved products are subject to these requirements and ongoing review. For drugs and vaccines, these requirements include submissions of safety and other post-marketing information and reports, plasma donor testing, registration requirements, CGMP, requirements relating to potency and stability, quality control, quality assurance, restrictions on advertising and promotion, import and export restrictions and recordkeeping requirements. Requirements for medical devices are similar and include QMSR compliance, establishment registration and device listing; record keeping; restrictions on advertising and promotion; post-market surveillance and reporting of adverse events and certain malfunctions, and restrictions on import and export. In addition, various state laws require that companies that manufacture and/or distribute drug products within the state obtain and maintain a manufacturer or distributor license, as appropriate. Some states have similar requirements for devices. Because of the breadth of these laws, it is possible that some of our business activities could be subject to challenge under one or more of such laws.
42
Government regulators enforce CGMP, QMSR, and other requirements through periodic unannounced inspections of manufacturing facilities. The FDA is authorized to inspect domestic and foreign manufacturing facilities without prior notice at reasonable times and in a reasonable manner. Health Canada may conduct similar inspections of our domestic and foreign facilities where products offered and sold in Canada are produced, or related formulation and filling operations are conducted. The FDA, Health Canada, and other foreign regulatory agencies conduct periodic inspections of our facilities. Following several of these inspections, regulatory authorities have issued inspectional observations, some of which were significant, but all of which are being, or have been, addressed through corrective actions. If, in connection with any future inspection, regulatory authorities find that we are not in substantial compliance with all applicable requirements, or if they are not satisfied with the corrective actions we take, our regulators may undertake enforcement action against us, which may include:
•warning letters, untitled letters, and other communications;
•product seizure or withdrawal of the product from the market;
•restrictions on the marketing or manufacturing of a product;
•suspension or withdrawal of regulatory approvals or refusal to approve pending applications or other marketing submissions, or supplements to approved applications;
•fines or disgorgement of profits or revenue; and
•injunctions or the imposition of civil or criminal penalties.
Similar action may be taken against us should we fail to comply with regulatory requirements, or later discover previously unknown problems with our products or manufacturing processes. For instance, our products are tested regularly to determine if they satisfy potency and stability requirements for their required shelf lives. Failure to meet potency, stability or other specification requirements could result in delays in distributions, recalls or other consequences.
Even if regulatory approval, clearance, or other marketing authorization of a product is granted, the approval, clearance, or marketing authorization may be subject to limitations on the indicated uses for which the product may be marketed or sold or to the conditions of approval. Regulatory approval or other authorization may also contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. If we experience any of these post-approval events, our business, financial condition, operating results and cash flows could be materially and adversely affected.
Failure to obtain or maintain regulatory approval in international jurisdictions could prevent us from marketing our products abroad and could limit the growth of our business.
We currently sell certain of our products outside the United States and intend to expand the countries in which we sell our products. We currently have market authorization under the mutual recognition procedure to sell BioThrax® in France and Germany. To market or sell our products in foreign jurisdictions under normal circumstances, we generally need to obtain separate regulatory approvals and comply with numerous and varying requirements or use alternative “emergency use” or other exemptions from general approval and import requirements. Approval by the FDA in the United States, in Canada by Health Canada, or by any individual EU member state does not ensure approval by all foreign regulatory authorities. The approval procedures in foreign jurisdictions can vary widely and can involve additional clinical trials and data review beyond that required by the FDA or any other global health authority. There is also a risk that a regulatory authority in another country could conclude that we have violated the rules and regulations related to product development, approval or promotion in that country. Therefore, there is a risk that we could be subject to a foreign enforcement action if found to be in violation of such laws and regulations. We and our collaborators may not be able to obtain foreign regulatory approvals on a timely basis, if at all, and we may be unable to successfully commercialize our products in desired jurisdictions internationally if no alternate procurement pathway is identified for authorized government customers in a particular jurisdiction. We have limited experience in preparing, filing and procuring the applications necessary to gain foreign regulatory approvals in all global jurisdiction and expect to rely on third-party contract research organizations and consultants to assist us in this process. Our reliance on third parties can introduce additional uncertainty into the process.
43
Laws and regulations governing international operations may preclude us from developing, manufacturing and selling certain products outside of the United States, require us to develop and implement costly compliance programs, and if violated, can lead to financial and other impacts.
As we continue to expand our activities outside of the United States, we are subject to an increased risk of violating, and must dedicate additional resources towards avoiding inadvertently conducting activities in a manner that violates, the U.S. Foreign Corrupt Practices Act (the "FCPA"), the U.K. Bribery Act, Canada's Corruption of Foreign Public Officials Act, and other similar foreign anti-bribery laws that prohibit corporations and individuals from corruptly paying, offering to pay, or authorizing the payment of anything of value, directly or indirectly, to any foreign government official, government staff member, political party or party official, or political candidate in an attempt to influence a person working in an official capacity or otherwise obtain an improper advantage. We may be held responsible for conduct of the third parties with which we interact. The FCPA also obligates companies whose securities are listed in the United States to comply with certain accounting provisions requiring the Company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations. Some anti-bribery laws also apply to private sector bribery. Compliance with the FCPA and other anti-bribery laws is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, the FCPA presents particular challenges in the pharmaceutical industry, because, in many countries, hospitals and other parts of the health system are operated by the government, and doctors, hospital employees, and other health care providers are considered foreign officials. Certain payments to hospital employees and other health care professionals in connection with clinical trials and other work have been deemed to be improper payments to government officials and have led to FCPA enforcement actions.
Many countries, including the United States, also have various lobbying laws and regulations governing the conduct of individuals and companies who interact with government officials. These laws and regulations typically include certain restrictions and disclosure obligations. If we, our employees, or third parties acting on our behalf do not comply with these laws and regulations, we may be subject to civil and criminal penalties.
Many countries, including the United States, restrict the export or import of products to or from certain countries through, for example, bans, sanction programs, and boycotts. Such restrictions may preclude us from supplying products in certain countries, which could limit our growth potential. Furthermore, if we, or third parties acting on our behalf, do not comply with these restrictions, we may be subject to civil and criminal penalties.
Various laws, regulations and executive orders also restrict the use and dissemination outside of the United States, or the sharing with certain non-U.S. nationals, of information classified for national security purposes, as well as certain products and technical data relating to those products. If we continue to expand our presence outside of the United States, it will require us to dedicate additional resources to comply with these laws, and these laws may preclude us from developing, manufacturing, or selling certain products and product candidates outside of the United States, which could limit our growth potential and increase our development costs. In addition, actions that the U.S. government and other governments have taken or threatened to take regarding tariffs and trade, and the associated uncertainty of how such actions may be implemented, may have adverse effects on the global economic environment and could also amplify these risks.
The failure to comply with laws governing international business practices may result in substantial civil and criminal penalties, suspension or debarment from government contracting, and other sanctions, and can cause reputational harm. The SEC also may bring enforcement actions against issuers for violations of the FCPA’s accounting provisions.
Changes to tax legislation may adversely affect our business.
In July 2025, the One Big Beautiful Bill Act (“OBBBA”) was signed into law, introducing significant tax changes. The OBBBA extends or makes permanent various tax provisions that were originally enacted in the 2017 Tax Cuts and Jobs Act and were set to expire at the end of 2025. The OBBBA features modified versions of individual and business tax relief proposals, and other new tax relief measures. In addition, it includes various revenue-raising measures, including changes to certain Inflation Reduction Act clean energy tax credits and various limits on business and individual tax deductions, that are intended to offset part of the cost of the legislation. These impacts did not have a material effect on our effective tax rate for the year ended December 31, 2025.
44
COMPETITIVE AND POLITICAL RISKS
Development and commercialization of pharmaceutical products, including for PHT preparedness, are routinely subject to evolving private and public sector competition.
The development and commercialization of new biopharmaceutical and medical technology products is highly competitive and subject to rapid technological advances. We will continue to face future competition from other companies and governments, universities and other non-profit research organizations in respect of our products, any products that we acquire, our current product candidates and any products we may seek to develop or commercialize in the future. The market for products can be subject to development of safer, more effective, more convenient or less costly products. The market for current products can also depend on what resources can be devoted to marketing or selling products, or how companies are positioned to adapt more quickly to new technologies, respond to scientific advances or patient preferences and needs, initiate or withstand substantial price competition and/or procure third-party licensing and collaborative arrangements.
There are a number of companies with products or product candidates addressing PHT preparedness that are competing with us for both USG procurement and development resources. Factors to consider include competitors' financial, technical, marketing and selling resources as well as potential leverage that their intellectual property estates may offer.
Any reduction in demand for our products or reduction or loss of development funding for our products or product candidates in favor of a competing product could lead to a loss of market share for our products and cause reduced revenues, margins and levels of profitability for us, which could adversely affect our business, financial condition, operating results and cash flows.
Our biologic products may face risks of competition from biosimilar manufacturers.
Biological products and product candidates, which we refer to as “Biologic Products,” can be affected by the approval and entry of “biosimilars” in the United States and other jurisdictions. Biosimilar products are licensed through an abbreviated pathway based on a showing that they are “highly similar” to a previously licensed product (known as the reference product) notwithstanding minor differences in clinically inactive components, and there are no clinically meaningful differences from the reference product in terms of safety, purity, and potency. Biologic Products in our current pipeline include CYFENDUS®, BioThrax® and ACAM2000®. If a biosimilar version of one of our Biologic Products were approved, it could have a material adverse effect on the sales and gross profits of the affected Biologic Product and could adversely affect our business, financial condition, operating results and cash flows.
NARCAN® (naloxone HCl) Nasal Spray is currently subject to generic and branded competition and may be subject to additional generic and branded competition in the future. If demand for over-the-counter NARCAN® Nasal Spray outpaces current estimations, there could be supply challenges to meet demand.
NARCAN® Nasal Spray was approved as an over-the-counter (“OTC”) medication in the U.S. in March 2023 and became available to retailers and e-commerce providers nationwide in August 2023. If demand for NARCAN® Nasal Spray increases beyond our current estimates, there could be supply interruptions. Although we have contingency plans to continue to provide product to those at the highest need and increase production to meet the anticipated increase in demand, such contingency plans may be unsuccessful or the implementation of such plans delayed, which could cause supply interruptions and adversely affect our business, financial condition, operating results and cash flows,
NARCAN® Nasal Spray currently faces generic competition. In 2016, Teva Pharmaceuticals Industries Limited and Teva Pharmaceuticals USA (collectively, “Teva”) filed an Abbreviated New Drug Application (an “ANDA”) seeking regulatory approval to market a generic version of NARCAN® Nasal Spray. In patent litigation related to Teva’s ANDA filing, a trial court decided in favor of Teva, and this decision was subsequently affirmed by the Court of Appeals for the Federal Circuit. The FDA approved Teva's ANDA in April 2019 and in December 2021 Teva commenced the launch of its generic naloxone nasal spray. As part of state settlements, including in Florida, Texas, Rhode Island, and West Virginia, Teva has agreed to supply Medication- Assisted Treatment (“MAT”) and generic opioid overdose reversal agents, like naloxone, to states at no cost in lieu of additional monetary compensation. The terms of these product donation agreements stretch 10 to 15 years. NARCAN® Nasal Spray also faces generic competition from Padagis LLC (“Padagis”) and Amneal Pharmaceuticals, Inc. (“Amneal”).
Sales of generic versions of NARCAN® Nasal Spray at prices lower than our branded product or provided at no cost by Teva, Padagis and Amneal have the potential to erode our sales and could impact our product revenue related to NARCAN® Nasal Spray.
NARCAN® Nasal Spray also faces branded competition from prescription products, such as Zimhi™ (naloxone), a branded injectable product developed by Adamis Pharmaceuticals Corporation, RextovyTM, (naloxone HCL nasal spray 4 mg), a branded product marketed by ZMI Pharma, Inc., Teleflex Medical Inc.'s Intranasal Mucosal Atomization Device, and Rezenopy® (naloxone HCL nasal spray 10mg), a branded product manufactured by Summit Biosciences Inc., as well as OTC products, such as RiViveTM (naloxene HCl nasal spray 3mg), a branded product developed by Harm Reduction Therapeutics.
45
NARCAN® (naloxone HCl) Nasal Spray may also face additional generic and branded competition in the future.
Political or social factors may delay or impair our ability to market and sell our products and may require us to spend significant management time and financial resources to address these issues.
Products developed to counter the potential impact of PHTs are subject to changing political and social environments. The political responses and social awareness of the risks of these threats on military personnel or civilians and the level of emphasis placed on such risks by the USG may vary over time. If the threat of terrorism were to decline, then the public perception of the risk on public health and safety may be reduced. This perception, as well as political or social pressures (including as a result of negative publicity we have received based on our longstanding ties to the USG), could delay or cause resistance to bringing our products in development to market or limit pricing or purchases of our products, any of which could negatively affect our revenues and our business, financial condition, operating results and cash flows.
In addition, substantial delays or cancellations of purchases could result from protests or challenges from third parties. Lawsuits brought against us by third parties or activists, even if not successful, could require us to spend significant management time and financial resources defending the related litigation and could potentially damage the public's perception of us and our products. Any publicity campaigns or other negative publicity may adversely affect the degree of market acceptance of our MCMs and thereby limit the demand for our products, which would adversely affect our business, financial condition, operating results and cash flows.
We may not be able to obtain orphan drug exclusivity for product candidates we may develop, and even if we do, that exclusivity may not prevent the FDA or foreign regulatory authorities from approving other competing products.
Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is a drug or biologic intended to treat a rare disease or condition. Generally, if a product candidate with an orphan drug designation subsequently receives the first marketing approval for the indication for which it has such designation, the product is entitled to a period of marketing exclusivity, which precludes the FDA from approving another marketing application for the same drug for the same indication for that time period. The applicable period is seven years in the United States.
In order for the FDA to grant orphan drug designation to one of our products, the agency must find, among other requirements, that the product is being or will be investigated for a condition or disease with a patient population of fewer than 200,000 individuals in the United States, or, for a vaccine, diagnostic drug, or preventive drug, it will be administered to fewer than 200,000 persons per year in the United States. Alternatively, the FDA may determine that there is no reasonable expectation that the costs of research and development of the drug can be recovered from sales of the drug in the United States. The FDA may conclude that the condition or disease for which we seek orphan drug designation does not meet this standard. Even if we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different products can be approved for the same indication. In addition, even after a product receives orphan drug exclusivity, the FDA can subsequently approve the same product for the same indication if the FDA or such authorities conclude that the later product is clinically superior in that it is shown to be safer, more effective or makes a major contribution to patient care; if the FDA determines that the holder of orphan drug exclusivity cannot ensure the availability of sufficient quantities of the product to meet the needs of patients with the rare disease or condition; or if the holder of orphan drug exclusivity consents to the approval of such subsequent product. Additionally, the FDA may revoke orphan drug designation if the FDA determines that the request for designation contained an untrue statement of material fact, omitted material information, or the FDA subsequently finds that the drug in fact had not been eligible for orphan drug designation at the time of submission of the request for designation.
We face similar risks in the EU and other foreign jurisdictions that have comparable regulations concerning orphan drug exclusivity.
INTELLECTUAL PROPERTY RISKS
Protection of our intellectual property rights is an important tool for sustaining our business and the failure to do so could impact our financial condition, operating results, and cash flows.
We actively seek to protect intellectual property rights related to our Company's assets, including patent rights, trademark rights, trade secrets, know-how and proprietary confidential information, through defense and enforcement of existing rights and pursuit of protection on new and arising innovations.
Obtaining, maintaining and enforcing our intellectual property rights in the United States and other countries remains a critical component of the development and commercialization of our Company's assets.
46
Some of the risks associated with procurement, maintenance and enforcement of intellectual property rights include changes in patent laws or administrative patent office rules, evolving criteria and eligibility of obtaining patent protection on particular subject matter, the validity and enforceability of our intellectual property rights, the potential scope of coverage of our intellectual property rights, and/or the availability or strength of legal remedies in a particular country to defend and enforce intellectual property rights.
Other risks include associated costs, such as costs of patent prosecution and maintenance and costs associated with post-grant challenges. For example, such costs include inter partes review proceedings in the United States and oppositions in Europe, as well as costs associated with litigating and enforcing patent and trademark rights.
Additional risks include limitations on our extent or ability to procure, maintain or defend intellectual property rights associated with in-licensed or acquired intellectual property, where, for example, other parties (e.g., licensors) may have the first right to maintain or defend intellectual property rights in which we have an interest, or may pursue strategies that are divergent to the interest of our Company.
Third-party claims of alleged patent infringement could delay, stop or otherwise affect the development and commercialization of our products and product candidates. Such challenges, while ongoing, could be costly, requiring and utilizing company resources. Such challenges, if successful, may impact marketing or launch of products, or require ongoing license and/or royalty fees associated with potential settlement agreements. These challenges may have the potential to materially harm our business, financial condition, operating results, and cash flows.
We are a party to a number of license agreements and expect to enter into additional license agreements in the future. Such license agreements or collaboration arrangements can be subject to challenges if interests or expectations under such license agreements diverge. Such challenges may be costly, risk time and resources, and could delay or impact development, commercialization or launch of our products.
Potential loss of proprietary information and know-how generally carries the risk of reducing the value of our technology and products.
We also rely upon unpatented proprietary technology, processes, and know-how, particularly as to our proprietary manufacturing processes. These types of proprietary confidential information, know-how, and trade secrets can be difficult to protect, and potential loss or misappropriation of this information generally carries the risk of reducing the value of our technology and products. We seek to protect this confidential information, in part, through agreements with our employees, consultants, and third parties, as well as through internal policies and audits, although these may not always be successful in protecting our proprietary confidential information, know-how, and trade secrets.
Certain of our products are approved as drug products under the provisions of the FDCA, which may render them susceptible to potential competition from generic manufacturers via the Hatch-Waxman Act and ANDA process. Other of our products may be susceptible to challenges by entry of biosimilars through the route established under the Biologics Price Competition and Innovation Act of 2009.
Although we intend to vigorously enforce our intellectual property rights, there can be no assurance that we will prevail in our enforcement or defense of our intellectual property rights. Our existing patents could be invalidated, found unenforceable, or narrowed in scope. Our trademark and trade name rights and related registrations may be challenged, opposed, infringed, diluted, canceled, circumvented, declared generic or determined to be infringing on other marks.
RISKS RELATED TO RELIANCE ON OTHER PARTIES
The loss of any of our non-exclusive, sole-source or single source suppliers, a shortage of related supplies or an increase in the price of materials supplied to us could have an adverse effect on our business, financial condition and results of operations.
We purchase certain supplies used in our manufacturing processes from non-exclusive, or single sources due to quality considerations, costs or constraints resulting from regulatory requirements. We depend on certain single-source suppliers for key materials, API, excipients, components, devices, manufacturing and other services necessary to produce and release the majority of our products and certain product candidates. For example, we rely on a single-source supplier to provide us with Alhydrogel® in sufficient quantities to meet our needs to manufacture CYFENDUS® and BioThrax® vaccines and the specialty plasma in our hyperimmune specialty plasma products and certain ingredients for the ACAM2000® vaccine. We also rely on single-source suppliers for the materials necessary to produce NARCAN® (naloxone HCl) Nasal Spray, such as the naloxone active pharmaceutical ingredient and other excipients, along with the vial, stopper and device.
47
Where a particular single-source supply relationship is terminated, we may not be able to establish additional or replacement suppliers for certain components or materials quickly. This is largely due to the FDA approval system, which mandates validation of materials prior to use in our products and product candidates, and the complex nature of manufacturing processes. In addition, we may lose a sole-source supplier due to, among other things, the acquisition of a supplier by a competitor (which may cause the supplier to stop selling its products to us) or the bankruptcy of such a supplier, which may cause the supplier to cease operations. Any reduction or interruption by a sole-source supplier of the supply of materials or key components used in the manufacturing of our products or product candidates, a reduction in quality or an increase in the price of those materials or components could adversely affect us. If we are unable to locate or establish alternative suppliers, our ability to manufacture our products and product candidates could be adversely affected and could harm our revenues, cause us to fail to satisfy contractual commitments, lead to a termination of one or more of our contracts or lead to delays in our clinical trials, any of which could be costly to us and otherwise materially harm our business, financial condition, operating results and cash flows.
We depend on third parties to conduct many of our clinical and non-clinical trials. If these third parties do not perform as contractually required or as we expect, we may not be able to obtain regulatory approval for or commercialize our product candidates and, as a result, our business, financial condition, operating results and cash flows may suffer.
We depend on third parties, such as independent clinical investigators, contract research organizations and other third-party service providers, to conduct the clinical and non-clinical trials of our product candidates and expect to continue to do so. We rely heavily on these third parties for successful execution of our clinical and non-clinical trials, but do not exercise day-to-day control over their activities. Our reliance on these service providers does not relieve us of our regulatory responsibilities, including ensuring that our trials are conducted in accordance with good clinical practice regulations and the plan and protocols contained in the relevant regulatory application. In addition, these organizations may not complete these activities on our anticipated or desired timeframe. We also may experience unexpected cost increases that are beyond our control. Problems with the timeliness or quality of the work of a contract research organization or other third party may lead us to seek to terminate the relationship and use an alternative service provider, which may prove difficult, costly and result in a delay of our trials. Any delay in or inability to complete our trials could delay or prevent the development, approval and commercialization of our product candidates.
In certain cases, government entities and non-governmental organizations ("NGOs") conduct studies of our product candidates, and we may seek to rely on these studies in applying for marketing approval for certain of our product candidates. These government entities and NGOs have no obligation or commitment to us to conduct or complete any of these studies or clinical trials and may choose to discontinue these development efforts at any time. Furthermore, government entities depend on annual Congressional appropriations to fund their development efforts, which may not be approved.
If we are unable to obtain any necessary third-party services on acceptable terms or if these service providers do not successfully carry out their contractual duties or meet expected deadlines, our efforts to obtain regulatory approvals for our product candidates may be delayed or prevented.
LEGAL AND REPUTATIONAL RISKS
Our financial condition and operating results could be adversely impacted by unfavorable results of legal proceedings or government investigations.
We are subject to various claims, legal proceedings and government investigations that have not yet been fully resolved, including stockholder derivative and putative class action lawsuits, and new matters may arise in the future. In addition, agreements entered into by us sometimes include indemnification provisions which can subject us to costs and damages in the event of a claim against an indemnified third party. The number of claims, legal proceedings and government investigations involving us, and the alleged magnitude of such claims, proceedings and government investigations, has generally increased over time and may continue to increase. Certain of these actions include, and future actual or threatened legal actions may include, claims for substantial and indeterminate amounts of damages, or may result in other actions adverse to us.
48
For example, in 2021, multiple purported class action lawsuits were filed against us and certain of our current and former senior officers in the United States District Court for the District of Maryland seeking unspecified damages on behalf of a putative class of persons who purchased or otherwise acquired shares of our common stock during various date ranges. The complaints alleged, among other things, that we made materially false and misleading statements regarding our procedures and quality controls relating to vaccine production, in violation of federal securities laws. In February 2025, the Court granted final approval of a settlement between us and the lead plaintiffs. As another example, multiple stockholder derivative lawsuits were filed in The Court of Chancery of the State of Delaware and the United States District Court for the District of Maryland on behalf of the Company against certain current and former officers and directors for breach of fiduciary duties, waste of corporate assets, unjust enrichment and insider trading, each allegation related to the Company’s capabilities to manufacture COVID-19 vaccine bulk drug substance. In addition to monetary damages, the complaints sought the implementation of multiple corporate governance and internal policy changes. In August 2025, the United States District Court for the District of Maryland granted final approval of a settlement with regard to the derivative matters.
Regardless of merit, litigation can be both time-consuming and disruptive to our operations and cause significant expense and diversion of management’s attention. The outcome of litigation or government investigations is also inherently uncertain. If one or more legal matters were resolved against us or an indemnified third party in a reporting period for amounts above management’s expectations, our financial condition and operating results for that reporting period could be materially adversely affected. Further, such an outcome could result in significant compensatory, punitive or trebled monetary damages, disgorgement of revenue or profits, remedial corporate measures or injunctive relief against us and could require us to change our business practices or limit our ability to offer certain products and services, all of which could materially adversely affect our financial condition and operating results. While we maintain insurance coverage for certain types of claims, such insurance coverage may be insufficient to cover all losses or all types of claims that may arise.
We rely significantly on information technology systems and any cyber-security incidents, unauthorized access or other failure, inadequacy, interruption or security lapse of that technology could harm our ability to operate our business effectively or result in data leakage of proprietary or confidential business or employee information.
Our business is increasingly dependent on critical, complex and interdependent information technology systems, including Internet-based systems, to support business processes as well as internal and external communications. We previously contracted with the USG and pharmaceutical companies for the development and manufacture of a significant quantity of COVID-19 vaccines, which raised our security profile and heightened potential risks that malicious actors may seek to disrupt our systems or misappropriate our information. The size and complexity of our computer systems and those of many of our business partners, collaborators and other third parties make them potentially vulnerable to interruption, invasion, computer viruses, destruction, unauthorized or malicious intrusion and additional related disruptions, which may result in the impairment of production and key business processes. Our systems and information are also potentially vulnerable to cybersecurity incidents through user error, phishing scams, or malfeasance, as well as cyber-security incidents involving our employees, business partners, collaborators or other third parties, any of which may expose sensitive data to unauthorized persons. Our systems and those of our business partners and collaborators have in the past been, and in the future likely will be subject to computer viruses, malicious codes, unauthorized access and other cybersecurity incidents. We are not aware of any significant impact on our operations or financial results from such incidents.
No system of protection is adequate to protect against all such threats, even if they are deemed to be industry standard, and there can be no assurance that we will be able to repel any such attacks. Cybersecurity incidents could lead to the loss of trade secrets or other intellectual property or the public exposure of personal information, including sensitive personal information, of our employees, clinical trial patients, customers and others. Evaluating and responding to any such threats may also be expensive and time-consuming. Any such unauthorized access to our information, whether through an incident involving our information technology systems or those of our business partners, collaborators or other third parties, could disrupt our business operations, result in the loss of assets, and have a material adverse effect on our reputation, business, financial condition, or results of operations. While the Company has experienced non-material cyber incidents involving third-party vendors, the Company’s continued use of third parties in its business yields the potential for material cybersecurity incidents that may harm business operations.
A significant business disruption or a breach in security resulting in misappropriation, theft or sabotage with respect to proprietary or confidential business or employee information could result in significant financial losses, legal, business or reputational harm to us, compromise our business prospects and our commitments to the USG or other customers, any of which could materially and adversely affect our business, financial condition and operating results.
We face product liability exposure, which could cause us to incur substantial liabilities and negatively affect our business, financial condition and results of operations.
We face an inherent risk of product liability exposure related to the sale of our products, any other products that we successfully acquire or develop and the testing of our product candidates in clinical trials.
49
One measure of protection against such lawsuits is coverage under the PREP Act which creates liability protection for manufacturers of biodefense countermeasures when the Secretary of HHS issues a declaration for their manufacture, administration or use. A PREP Act declaration is meant to provide liability protection from all claims under federal or state law for loss arising out of the administration or use of a covered countermeasure under a government contract. The Secretary of HHS has issued PREP Act declarations covering countermeasures for smallpox, mpox, and other orthopox; anthrax; and botulinum toxin. These declarations apply to certain of our products, namely BioThrax®, ACAM2000®, CYFENDUS®, raxibacumab, ANTHRASIL®, BAT® and CNJ-016® (VIGIV) products, as covered countermeasures. Manufacturers are not entitled to protection under the PREP Act in cases of willful misconduct or for cases brought in non-U.S. tribunals or under non-U.S. law. We cannot predict whether the Secretary of HHS will renew the declarations when they expire, whether Congress will fund the relevant PREP Act compensation programs, or whether the necessary prerequisites for immunity would be triggered with respect to our products or product candidates.
Additionally, certain of our products, namely BioThrax®, are under the SAFETY Act, which provides certain product liability limitations for qualifying anti-terrorism technologies for claims arising from or related to an act of terrorism. Although BioThrax® is designated and certified under the SAFETY Act, the law may not provide adequate protection from claims made against us.
If we cannot successfully defend ourselves against future claims that our products or product candidates caused injuries and if we are not entitled to indemnity by the USG, or the USG does not honor its obligations to us under the PREP Act or SAFETY Act, or if the liability protections under the PREP Act and SAFETY Act are not adequate to cover all claims, we may incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
•decreased demand or withdrawal of a product;
•injury to our reputation;
•withdrawal of clinical trial participants;
•costs to defend the related litigation;
•substantial monetary awards to trial participants or patients;
•loss of revenue; and
•an inability to commercialize products that we may develop.
The amount of insurance that we currently hold may not be adequate to cover all liabilities that we may incur. Further product liability insurance may be difficult and expensive to obtain. We may not be able to maintain insurance coverage at a reasonable cost and we may not be able to obtain insurance coverage that will be adequate to satisfy all potential liabilities. For example, we may not have sufficient insurance against potential liabilities associated with a possible large-scale deployment of BioThrax® vaccine as a countermeasure to a bioterrorism threat. We rely on PREP Act protection for BioThrax®, raxibacumab, ACAM2000®, CYFENDUS®, ANTHRASIL®, BAT® and CNJ-016® (VIGIV) products, and SAFETY Act protection for BioThrax® in addition to our insurance coverage to help mitigate our product liability exposure for these products. Additionally, potential product liability claims related to our commercial products may be made by patients, health care providers or others who sell or consume these products. Such claims may be made even with respect to those products that possess regulatory approval for commercial sale. Claims or losses in excess of our product liability insurance coverage could have a material adverse effect on our business, financial condition, operating results and cash flows.
FINANCIAL RISKS
Our level of indebtedness and the terms of our indebtedness could adversely affect our business and liquidity position. We may not have sufficient cash flow from our operations to pay our substantial debt.
As described in Note 11, “Debt” in the Notes to Consolidated Financial Statements in Part II, Item 8. of this Annual Report on Form 10-K, as of December 31, 2025, we have approximately $589.7 million of total indebtedness, which includes our outstanding Senior Unsecured Notes.
Our level of indebtedness could have important consequences for us, including:
•limiting our ability to obtain additional financing, if needed, for working capital, capital expenditures, acquisitions, debt service requirements or other purposes;
•increasing our vulnerability to adverse economic, industry or competitive developments;
•limiting our flexibility in planning for, or reacting to, changes in our business and industry; and
•placing us at a competitive disadvantage compared to our competitors with less debt.
50
Our indebtedness may increase from time to time for various reasons, including fluctuations in operating results, working capital needs, capital expenditures, acquisitions and/or joint ventures. The cost and level of our debt could negatively impact our liquidity, future financing costs and financial results, while potential credit rating downgrades or adverse market conditions could increase borrowing costs or limit access to capital. Our cash flow and capital resources may not be sufficient to meet our debt obligations, and alternative financing measures may not be available on terms that are acceptable to us, or at all.
Our ability to service or refinance our debt depends on our future performance, which is subject to economic, financial, competitive and other factors beyond our control. We may also seek additional debt financing to support our ongoing activities or to provide additional financial flexibility. Debt financing can have significant adverse consequences for our business, including:
•requiring us to dedicate a substantial portion of cash flows from operations to payment on our debt, which would reduce available funds for other corporate initiatives;
•increasing the amount of interest that we have to pay on debt with variable interest rates, if market rates of interest increase, to the extent we are unable to offset such risk through our hedging instruments;
•subjecting us, as under our Senior Secured Credit Facilities and the indenture governing the Senior Unsecured Notes, to restrictive covenants that reduce our ability to take certain corporate actions, acquire companies, products or technology, or obtain further debt financing;
•requiring us to pledge our assets as collateral, which could reduce financial flexibility; and
•increasing our exposure to adverse economic and industry conditions, furthering disadvantaging us against our competitors that have less debt, better debt servicing options or stronger debt servicing capacity.
In addition, failure to comply with the covenants under our Senior Secured Credit Facilities and other debt agreements, including the maintenance of a specified gross leverage ratio, fixed charge coverage ratio and minimum liquidity level, could result in an event of default. An event of default could result in the acceleration of amounts due under a particular debt agreement and a cross-default and acceleration under other debt agreements. If such events occur, we may not have sufficient funds to pay or financing options to meet these obligations.
Our current indebtedness restricts and any additional debt financing may restrict the operation of our business and limit the cash available for investment in our business operations.
The Senior Secured Credit Facilities include the Term Loan Facility, which had an outstanding principal balance of $150.0 million as of December 31, 2025, and the ability to borrow up to $100.0 million under the Revolving Credit Agreement (subject to certain adjustments described therein). In addition, we have outstanding an aggregate principal amount of $439.7 million of our Senior Unsecured Notes. We may also seek additional debt financing to support our ongoing activities or to provide additional financial flexibility. Debt financing can have significant adverse consequences for our business, including:
•the level, timing and cost of product sales and Bioservices;
•the extent to which we acquire or invest in and integrate companies, businesses, products or technologies;
•the acquisition of new facilities and capital improvements to new or existing facilities;
•the payment obligations under our indebtedness;
•the scope, progress, results and costs of our development activities;
•our ability to obtain funding from collaborative partners, government entities and non-governmental organizations for our development programs;
•limiting the extent to which we may repurchase shares of our common stock; and
•the costs of commercialization activities, including product marketing, sales and distribution.
In addition, our Senior Secured Credit Facilities and our Senior Unsecured Notes each contain cross-default provisions whereby a default under one agreement would likely result in cross defaults under agreements covering other indebtedness. For example, if we default under the Senior Secured Credit Facilities, the lenders would have the right to accelerate the repayment of borrowings under the Senior Secured Credit Facilities, which would result in a cross-default and acceleration of the Company’s obligations under the Senior Unsecured Notes. The occurrence of a default under any of these arrangements would permit the holders of the notes or the lenders under our Senior Secured Credit Facilities to declare all amounts outstanding under those borrowing arrangements to be immediately due and payable, and there is no assurance that we would have sufficient funds to satisfy any such accelerated obligations.
51
Our hedging programs have been, and any hedging program we initiate in the future will be, subject to counterparty default risk.
From time to time, we manage our interest rate risk in part by entering into interest rate swaps with a number of counterparties to swap a portion of our indebtedness that is based on variable interest rates to a fixed rate. As a result, when we are party to such interest rate swaps, we are subject to the risk that the counterparty to one or more of these contracts defaults on its performance under the contract. During an economic downturn, the counterparty's financial condition may deteriorate rapidly and with little notice and we may be unable to take action to protect our exposure. In the event of a counterparty default, we could incur losses, which may harm our business and financial condition. In the event that one or more of our counterparties becomes insolvent or files for bankruptcy, our ability to eventually recover any losses suffered as a result of that counterparty's default may be limited by the liquidity of the counterparty.
We may be unable to continue to progress on or implement our strategic plans and sustain our current operating performance, in which case our business, results of operations, financial condition and prospects could be adversely affected, and which may give rise to substantial doubt regarding our ability to continue as a going concern.
As of December 31, 2025, we had unrestricted cash and cash equivalents of $205.4 million and remaining capacity under the Revolving Credit Agreement of $100.0 million. Also as of December 31, 2025, we had borrowings of $150.0 million on our Term Loan Facility and $439.7 million of Senior Unsecured Notes outstanding. The Company may be unable to comply with debt covenants in future periods without additional sources of liquidity.
If our capital resources are insufficient to meet our future capital requirements, we will need to finance our cash needs through public or private equity or debt offerings, bank loans or collaboration and licensing arrangements. In August 2025, we filed a shelf registration statement on Form S-3, which was declared effective on August 15, 2025 (the "Shelf Registration Statement"). The Shelf Registration Statement allows us to sell from time to time up to $250 million of common stock, preferred stock, warrants, debt securities, depositary shares, rights to purchase common stock and units that include any of these securities.
If we raise funds by issuing equity securities, including through a registrations statement, our stockholders may experience dilution. Debt financing, if available, may involve agreements that include covenants, like those contained in our Senior Secured Credit Facilities and the indenture governing the Senior Unsecured Notes, limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures, pursuing acquisition opportunities or declaring dividends. If we raise funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish valuable rights to our technologies or product candidates or grant licenses on terms that may not be favorable to us. Our Senior Secured Credit Facilities as well as the indenture governing the Senior Unsecured Notes may restrict our ability to incur additional indebtedness.
Economic conditions may make it difficult to obtain financing on attractive terms, or at all. If financing is unavailable or lost, our business, operating results, financial condition and cash flows would be adversely affected, and we could be forced to delay, reduce the scope of or eliminate many of our planned activities.
We may not maintain profitability in future periods or on a consistent basis.
Our profitability has been substantially dependent on product sales, which historically have fluctuated significantly from quarter to quarter, and we expect that they will continue to fluctuate significantly based primarily on the timing of our fulfillment of orders from the USG. We may not be able to achieve consistent profitability on a quarterly basis or sustain or increase profitability on an annual basis.
Impairment charges to our intangible assets or property, plant and equipment could have a material adverse effect on our business, results of operations and financial condition.
In accordance with GAAP, we are required to assess the value of our intangible assets and goodwill annually, or more frequently whenever events or changes in circumstances indicate potential impairment, such as changing market conditions or any changes in key assumptions. If the testing performed indicates that an asset may not be recoverable, we are required to record a non-cash impairment charge for the difference between the carrying value of the asset and its implied fair value in the period the determination is made.
We also periodically monitor the remaining net book values of our property, plant and equipment, or whenever events or changes in circumstances indicate that the carrying amount of an asset group may not be recoverable.
52
We have a significant amount of intangible assets and property, plant and equipment on our balance sheet and in the past have been required to recognize non-cash impairment charges. The impairment tests require us to make an estimate of the fair value of our reporting units. An impairment could be recorded as a result of changes in assumptions, estimates or circumstances, some of which are beyond our control. Since a number of factors may influence determinations of fair value, we are unable to predict whether impairments of intangible assets and property, plant and equipment will occur in the future, and we can provide no assurance that continued conditions will not result in future impairments of these assets. The future occurrence of a potential indicator of impairment could include matters such as (i) a decrease in expected net earnings, (ii) adverse equity market conditions, (iii) a decline in current market multiples, (iv) a decline in our common stock price, (v) a significant adverse change in legal factors or the general business climate, and (vi) an adverse action or assessment by a regulator. Any such impairment would result in us recognizing a non-cash charge in our Consolidated Balance Sheets, which could adversely affect our business, results of operations and financial condition.
The accuracy of our financial reporting depends on the effectiveness of our internal control over financial reporting. Any material weakness in our internal control over financial reporting could have an adverse effect on our business and financial results and our ability to meet our reporting obligations could be negatively affected, each of which could negatively affect the trading price of our common stock.
Internal control over financial reporting can provide only reasonable assurance with respect to the preparation and fair presentation of financial statements and may not prevent or detect misstatements. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis. Failure to maintain effective internal control over financial reporting, or lapses in disclosure controls and procedures, could impact our financial information and disclosures, require significant resources to remediate, and expose us to legal or regulatory proceedings.
We regularly review and update our internal controls and disclosure controls and procedures. In addition, we are required under the Sarbanes-Oxley Act of 2002 to report annually on our internal control over financial reporting.
The expansion of our international operations increases our risk of exposure to credit losses.
As we continue to expand our business activities with foreign governments in certain countries that have experienced deterioration in credit and economic conditions or otherwise, our exposure to uncollectible accounts will rise. Global economic conditions and liquidity issues in certain countries have resulted and may continue to result in delays in the collection of accounts receivable and may result in credit losses. Future governmental actions and customer specific actions may require us to re-evaluate the collectability of our accounts receivable and we may potentially incur credit losses that materially impact our operating results.
RISKS RELATED TO STRATEGIC ACQUISITIONS, DIVESTITURES AND COLLABORATIONS
We may not be successful in identifying, structuring or acquiring businesses and products to drive our growth.
We may not be successful in identifying, effectively evaluating, structuring, acquiring or in-licensing, and developing and commercializing additional products on favorable terms, or at all. Competition for attractive product opportunities is intense and may require us to devote substantial resources, both managerial and financial, to an acquisition opportunity. A number of more established companies are also pursuing strategies to acquire or in-license products in the biopharmaceutical field. These companies may have a competitive advantage over us due to their size, cash resources, cost of capital, effective tax rate or greater clinical development and commercialization capabilities.
Acquisition efforts can consume significant management attention and require substantial expenditures, which could detract from our other programs. In addition, we may devote significant resources to potential acquisitions that are never completed. Even if we are successful in acquiring a company or product, it may not result in a successfully developed or commercialized product or, even if an acquired product is commercialized, competing products or technologies could render a product noncompetitive, uneconomical or obsolete. Moreover, the cost of acquiring other companies or in-licensing products could be substantial, and in order to acquire companies or new products, we may need to incur substantial debt or issue dilutive securities.
If we are unsuccessful in our efforts to identify and acquire other companies, products, or in-license and develop additional products, or if we acquire or in-license unproductive assets, it could have a material adverse effect on the growth of our business, and we could be compelled to record significant impairment charges to write-down the carrying value of our acquired intangible assets, which could materially harm our business, financial condition, operating results and cash flows.
53
Our failure to successfully integrate acquired businesses and/or assets into our operations could adversely affect our ability to realize the benefits of such acquisitions and, therefore, to grow our business.
We may not be able to integrate any acquired business successfully or operate any acquired business profitably. In addition, cost synergies, if achieved at all, may be less than we expect, or may take greater time to achieve than we anticipate.
Issues that could delay or prevent successful integration or cost synergies of an acquired business or products include, among others:
•retaining existing customers and attracting new customers;
•retaining key employees;
•diversion of management attention and resources;
•conforming internal controls, policies and procedures, business cultures and compensation programs;
•consolidating corporate and administrative infrastructures;
•successfully executing technology transfers and obtaining required regulatory approvals;
•consolidating sales and marketing operations;
•identifying and eliminating redundant and underperforming operations and assets;
•assumption of known and unknown liabilities;
•coordinating geographically dispersed organizations;
•managing tax costs or inefficiencies associated with integrating operations; and
•risks associated with intellectual property rights related to an acquisition or collaboration, including but not limited to, license rights, freedom-to-operate, litigation, and loss of proprietary confidential information, know-how, and trade secrets.
If we are unable to successfully integrate pending and future acquisitions with our existing businesses, or operate any acquired business profitably, we may not obtain the advantages that the acquisitions were intended to create, which may materially adversely affect the growth of our business, financial condition, operating results and cash flows.
Divestitures and sales of assets could negatively impact our business, and retained liabilities from businesses or assets that we have sold could adversely affect our financial results.
In connection with the execution of our multi-year strategic plan to stabilize, turnaround and transform the Company, we have completed several divestitures and sales of assets. These divestitures and asset sales pose risks and challenges that could negatively impact our business, including retained liabilities related to divested businesses and sold assets, obligations to indemnify buyers against contingent liabilities and potential disputes with buyers.
If post-completion liabilities and obligations related to divestitures and asset sales are substantial and exceed our expectations, our financial position, results of operations and cash flows could be negatively impacted. Any divestiture or asset sale may result in a dilutive impact to our future earnings if we are unable to offset the dilutive impact from the loss of revenue and profits associated with the divestiture or sold asset, as well as significant write-offs, including those related to goodwill and other intangible assets, which could have a material adverse effect on our results of operations and financial condition.
54
RISKS RELATED TO OWNERSHIP OF OUR COMMON STOCK
Our business or our share price could be negatively affected as a result of the actions of stockholders.
In recent years, some stockholders have placed increasing pressure on publicly traded companies in our industry and others to effect changes to corporate governance practices, executive compensation practices, social and environmental practices, disclosures related to certain matters and to undertake certain corporate actions. Stockholders may advocate for companies to take varied, and at times conflicting, approaches toward these issues. In this dynamic landscape, we can provide no assurance that stockholders will not publicly advocate for us to make changes to one or more of these areas or to engage, or not to engage, in certain corporate actions. Responding to challenges from stockholders, such as stockholder proposals, media campaigns, proxy contests or other public or private means, could be costly and time consuming and could have an adverse effect on our reputation and divert the attention and resources of management and our board of directors, which could have an adverse effect on our business and operational results. Any such stockholder actions or requests, or the mere public presence of stockholders with a reputation for taking such actions among our investor base, could also cause the market price of our common stock to experience periods of increased volatility.
Provisions in our certificate of incorporation and by-laws and under Delaware law may discourage acquisition proposals, delay a change in control or prevent transactions that stockholders may consider favorable.
Provisions in our certificate of incorporation and by-laws may discourage, delay or prevent a merger, acquisition or other changes in control that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. These provisions may also prevent or frustrate attempts by our stockholders to replace or remove our management.
These provisions include:
•the classification of our directors;
•limitations on changing the size of our board of directors;
•limitations on the removal of directors;
•limitations on filling vacancies on the board of directors;
•advance notice requirements for stockholder nominations of candidates for election to the board of directors and other proposals to be voted on at meetings of stockholders;
•the inability of stockholders to act by written consent;
•the inability of stockholders to call special meetings; and
•the ability of our board of directors to designate the terms of and issue a new series of preferred stock without stockholder approval.
The affirmative vote of a majority of our board of directors or the holders of our capital stock representing at least 75% of the voting power of all outstanding stock entitled to vote is required to amend or repeal the above provisions of our certificate of incorporation or by-laws. The affirmative vote of either a majority of the directors present at a meeting of our board of directors or holders of our capital stock representing at least 75% of the voting power of all outstanding stock entitled to vote is required to amend or repeal our by-laws.
In addition, we are subject to Section 203 of the Delaware General Corporation Law ("Section 203"). In general and subject to certain exceptions, Section 203 prohibits a publicly-held corporation from engaging in a business combination with an interested stockholder, generally a person which, together with its affiliates, owns or within the last three years has owned 15% or more of the corporation's voting stock, for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner. Accordingly, Section 203 may discourage, delay or prevent a change in control of us.
Our board of directors may adopt a new stockholder rights plan without stockholder approval, which could prevent a change in control of us in instances in which some stockholders may believe a change in control is in their best interests.
Our board of directors may adopt a stockholder rights plan without stockholder approval, which may have anti-takeover effects, potentially preventing a change in control of us in instances in which some stockholders may believe a change in control is in their best interests. This could cause substantial dilution to a person or group that attempts to acquire us on terms that our board of directors does not believe are in our best interests or those of our stockholders and may discourage, delay or prevent a merger or acquisition that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares.
55
Our stock price is volatile, and purchasers of our common stock could incur substantial losses.
The price of our common stock has been, and is likely to continue to be, volatile. The market price of our common stock could fluctuate significantly for many reasons, including in response to the risks described in this “Risk Factors” section, or for reasons unrelated to our operations, such as reports by industry analysts, investor perceptions or negative announcements by our customers, competitors or suppliers regarding their own performance, as well as industry conditions and general financial, economic and political instability. From January 1, 2023 through December 31, 2025, our common stock traded as high as $16.66 per share and as low as $1.42 per share. The market price of our common stock may be influenced by many factors, including, among others:
•contracts, decisions and procurement policies by the USG, and the addition or loss of any other customer affecting our anthrax vaccines and our other products and product candidates;
•CDMO contracts with collaboration partners;
•the success of competitive products or technologies;
•results of clinical and non-clinical trials of our product candidates;
•announcements of acquisitions, financings or other transactions by us;
•current or future litigation, legal proceedings, or governmental investigations;
•regulatory or public concern as to the safety of our products;
•termination or delay of a development program;
•the recruitment or departure of key personnel;
•variations in our product revenue and profitability; and
•the other factors described in this “Risk Factors” section.
Because we currently do not pay dividends, investors will benefit from an investment in our common stock only if it appreciates in value.
We currently do not pay dividends on our common stock. Our Senior Secured Credit Facilities and the indenture governing our Senior Unsecured Notes limit and any future debt agreements that we enter into may limit our ability to pay dividends. As a result, capital appreciation, if any, of our common stock will be the sole source of gain for our stockholders based on current expectations.
Future sales of our common stock or other securities convertible into common stock, or the perception that such sales or issuances could occur, could result in dilution of our stockholders and could cause our share price to decline.
Our board of directors is authorized, without stockholder approval, to cause us to issue additional shares of our common stock or to raise capital through the issuance of preferred shares or the sale of debt securities that are convertible into common stock, options, warrants and other rights, on terms and for consideration as our board of directors in its sole discretion may determine. We may require or desire additional funding in the future and we may seek to achieve such funding through future sales of our common stock or other securities convertible into our common stock. Sales of substantial amounts of our common stock or the issuance of preferred shares, convertible debt, options, restricted stock units, performance stock units, warrants and other rights, or the perception that such sales or issuances could occur could cause the market price of our common stock to decrease significantly. As of December 31, 2025, we had 52,132,099 shares of common stock issued and outstanding. We cannot predict the effect, if any, of future sales of our common stock or any preferred shares, convertible debt securities, options, restricted stock units, performance stock units, warrants or other rights or the availability of our common stock for future sales on the value of our common stock.
56
We cannot guarantee that our 2025 Share Repurchase Program will be utilized in full, or at all, or that our 2025 Share Repurchase Program will enhance long-term stockholder value.
Our Board of Directors authorized the 2025 Share Repurchase Program for up to $50.0 million of the Company’s common stock through March 27, 2026, and through December 31, 2025 we repurchased approximately $25.1 million under the Share Repurchase Program. In February 2026, the Company reauthorized the 2025 Share Repurchase Program for the repurchase of up to $50.0 million of the Company's common stock through March 31, 2027. Any share repurchases will depend upon, among other factors, market conditions, the market price of the Company’s common shares, macroeconomic environment and other investment opportunities. The existence of the 2025 Share Repurchase Program could cause our stock price, in certain cases, to be higher or lower than it otherwise would be and could potentially reduce the market liquidity or have other unintended consequences for our stock. We can provide no assurance that we will repurchase shares of our common stock at favorable prices, if at all. Although the 2025 Share Repurchase Program is intended to enhance long-term stockholder value, we can provide no assurance it will do so. The 2025 Share Repurchase Program does not obligate the Company to acquire any particular amount of common stock and it may be suspended or discontinued, or the amount to be spent by the Company to repurchase shares could be reduced, at any time at the Company’s discretion. Any decision to reduce or discontinue repurchasing shares of our common stock pursuant to our 2025 Share Repurchase Program could cause the market price for our common stock to decline and may negatively impact our reputation and investor confidence in us.
GENERAL RISK FACTORS
Our success is dependent on our continued ability to attract, motivate and retain key personnel, and any failure to attract or retain key personnel may negatively affect our business.
Because of the specialized scientific nature of our business, our ability to develop products and to compete with our current and future competitors largely depends upon our ability to attract, retain and motivate highly qualified managerial and key scientific and technical personnel (including quality and manufacturing personnel). If we are unable to retain the services of one or more of the principal members of senior management or other key employees, our ability to implement our business strategy could be materially harmed. From time to time, there may be changes in our senior management team resulting from the hiring or departure of executives. In addition, we face intense competition for qualified employees from biopharmaceutical companies, research organizations and academic institutions. Attracting, retaining or replacing these personnel on acceptable terms may be difficult and time-consuming given the high demand in our industry for similar personnel. We believe part of being able to attract, motivate and retain personnel is our ability to offer a competitive compensation package, including equity incentive awards. If we cannot offer a competitive compensation package to attract and retain the qualified personnel necessary for the continued development of our business, we may not be able to maintain our operations or grow our business.
57
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not applicable.
ITEM 1C. CYBERSECURITY
CYBERSECURITY
The ERM program and ERA process is described in the Company’s Enterprise Risk Management Policy. The program includes enterprise level risks grouped in 12 risk categories. Cybersecurity is included as a standing risk category. The ERM program does not itself independently review cybersecurity policies and practices. ERM, in collaboration with Emergent’s Policy and Training Center of Excellence, provides training on Emergent’s Enterprise Risk Management Policy to all employees who are at the vice president level and above. As part of our ongoing ERM enhancements, our ERM intranet page was launched in 2024 to centralize risk-related resources and policies. The Company periodically performs a comprehensive reassessment of the Company's enterprise risks, with results thoroughly reviewed and communicated to executive leadership (VP and above) to align on key risks and strategic priorities. Annually, we provide an ERM training to all participants in advance of the Company's annual ERA. Full retraining on the ERM policy will occur every three years. The Company leverages the Committee of Sponsoring Organizations’("COSO") guidelines as the foundation for our ERM program and leverage external expertise.
The Company proactively reviews the threat landscape, impacts to the company, and addresses any gaps where necessary. Also, we maintain security operations metrics and incident response plan and conduct tabletop exercises. The Company engages outside consultants to review both its Cybersecurity posture and maturity, and to perform cyber assessments for the Company’s manufacturing/operational technology environments. The Company utilizes its Third-Party Risk Management Assessment Process to oversee and identify material risks from cybersecurity threats associated with its use of any third-party service provider. We utilize the NIST framework, which covers 23 categories. When applicable, we may inquire if the third-party vendor is SOC1/2, GDPR, certified. Additionally, the Company maintains cybersecurity insurance to help mitigate the impacts of potential cybersecurity incidents.
•Compliance with good (“x” = manufacturing, clinical, laboratory, pharmacovigilance, storage, distribution etc.) (GxP) and medical device Quality Management Systems Regulations (QMSR);
•Healthcare compliance, anti-corruption, privacy and data security landscape, medical product safety, supply chain, employee health and safety, political expenditures and lobbying activities, and government contracting;
•ERM program; and
•Cyber and information security risks.
58
59
ITEM 2. PROPERTIES
We own and lease approximately 1.0 million square feet of building space for development and manufacturing, laboratories, fill/finish facility services, offices and warehouse space for the conduct of our businesses at 11 locations in North America and Europe. Properties that have been leased expire on various dates between 2026 and 2034. Principal locations include:
Location | Use | Approximate square feet | Owned/leased | Operating Segment | ||||||||||
| Lansing, Michigan | Manufacturing operations, office and laboratory space. | 336,000 | Owned | Products & Services | ||||||||||
| Winnipeg, Manitoba, Canada | Manufacturing operations, office and laboratory space. | 160,000 (Owned); 15,800 (Leased) | Owned/Leased | Products & Services | ||||||||||
| Gaithersburg, Maryland | Laboratory space, office space and rental real estate. | 173,000 (Owned); 11,547 (Leased) | Owned/Leased | Products & Services | ||||||||||
| Canton, Massachusetts | Manufacturing operations and warehouse space. | 47,000 (Owned); 27,000 (Leased) | Owned/Leased | Products & Services | ||||||||||
| Elkridge, Maryland | Warehouse space. | 103,182 | Leased | Products & Services | ||||||||||
| Rockville, Maryland | Manufacturing facilities, office and warehouse space. | 84,295 | Owned | Products & Services | ||||||||||
Each property is considered to be in good condition, adequate for its purpose, and suitably utilized according to the individual nature and requirements of the relevant operations. Our policy is to improve and replace property as considered appropriate to meet the needs of the individual operations.
ITEM 3. LEGAL PROCEEDINGS
See Item 8 of Part II, “Financial Statements and Supplemental Data — Notes to Consolidated Financial Statements" — Note 20, "Litigation."
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable
60
PART II
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information and Holders
Our common stock trades on the New York Stock Exchange under the symbol "EBS".
As of February 19, 2026, the closing price per share of our common stock on the New York Stock Exchange was $10.99 and we had 46 holders of record of our common stock. This number does not include beneficial owners whose shares are held by nominees in street name.
Purchases of Equity
In March 2025, the Company announced that its Board of Directors authorized the repurchase of up to $50.0 million of the Company’s common stock (the “2025 Share Repurchase Program”) on or before March 27, 2026. In February 2026, the Company reauthorized the 2025 Share Repurchase Program for the repurchase of up to $50.0 million of the Company's common stock through March 31, 2027. Repurchases under the 2025 Share Repurchase Program may be made from time to time on the open market or in privately negotiated transactions. The timing and amount of any shares repurchased will be determined by the Company’s management based on its evaluation of market conditions and other factors, including the market price of the Company’s common stock, macroeconomic environment and other investment opportunities, consistent with applicable law. The 2025 Share Repurchase Program may be suspended or discontinued at any time.
The table below presents information regarding shares of our common stock that we repurchased during the three months ended December 31, 2025 under the 2025 Share Repurchase Program:
Issuer Purchases of Equity Securities | ||||||||||||||||||||||||||
Periods | Total Number of Shares Purchased | Average Price Paid Per Share (1) | Total Number of Shares Purchased as Part of Publicly Announced Plans or Programs | Approximate Dollar Value of Shares That May Yet Be Purchased Under the Plans or Programs (2) | ||||||||||||||||||||||
| October 1, 2025 - October 31, 2025 | 336,872 | $ | 9.45 | 336,872 | $ | 30,804,402 | ||||||||||||||||||||
| November 1, 2025 - November 30, 2025 | 272,921 | $ | 10.35 | 272,921 | $ | 27,945,048 | ||||||||||||||||||||
| December 1, 2025 - December 31, 2025 | 253,261 | $ | 11.93 | 253,261 | $ | 24,887,579 | ||||||||||||||||||||
Total | 863,054 | 863,054 | ||||||||||||||||||||||||
(1) Exclusive of commissions and nondeductible 1% excise tax, which excise was imposed pursuant to the Inflation Reduction Act of 2022. | ||||||||||||||||||||||||||
(2) Dollar amounts presented are inclusive of commissions and nondeductible 1% excise tax. | ||||||||||||||||||||||||||
Dividend Policy
We have not declared or paid any cash dividends on our common stock since becoming a publicly traded company in November 2006. We currently have no plans to pay dividends.
The remaining information required by Item 5 is hereby incorporated by reference from our Definitive Proxy Statement relating to our 2026 Annual Meeting of the Stockholders, to be filed with the SEC within 120 days following the end of our fiscal year.
61
Stock Performance Graph
The following graph provides a comparison of five year cumulative total stockholder returns of Emergent BioSolutions Inc.'s common stock, the Standard & Poor’s ("S&P") 500 Stock Index, the Russell 2000 Index, the S&P SmallCap 600 Index, the S&P Pharmaceuticals Index and the S&P Biotechnology Index. The annual changes for the five-year period shown on the graph are based on the assumptions that $100 had been invested in Emergent BioSolutions Inc.'s common stock and each index on December 31, 2020, all fiscal years end December 31st and all dividends were reinvested.
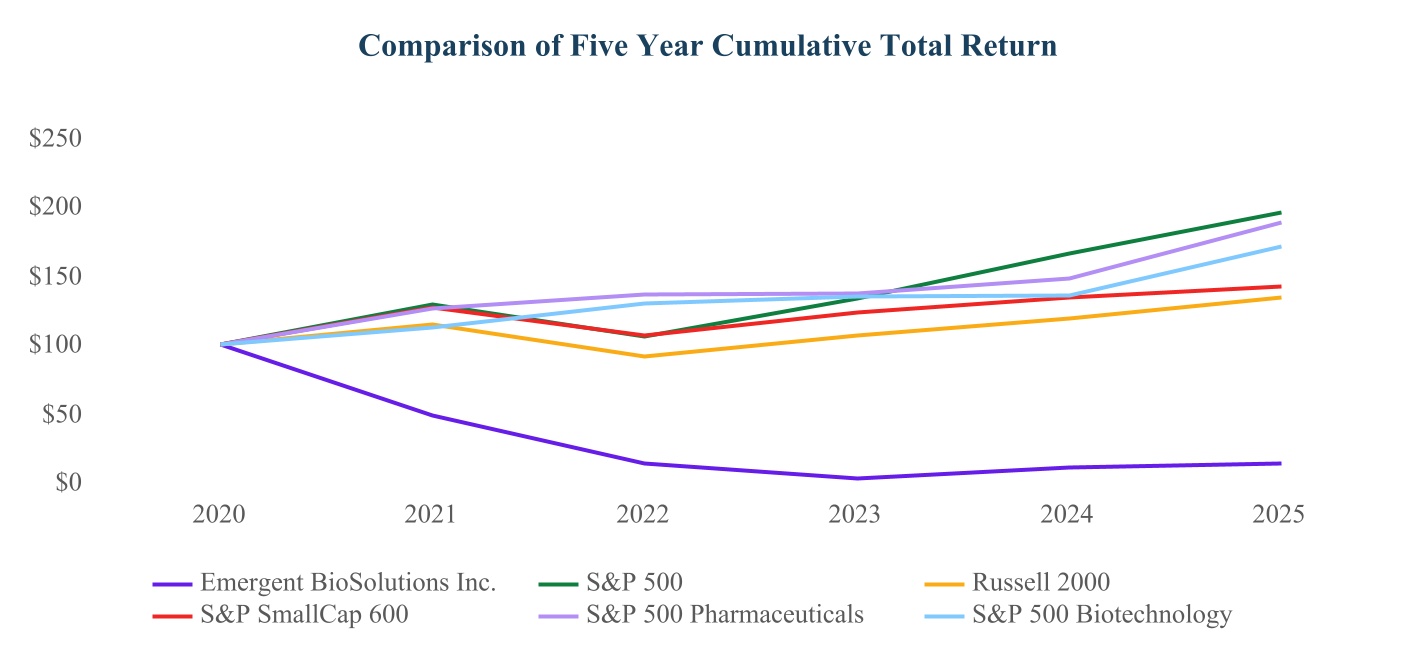
Market Performance | ||||||||||||||||||||
Company / Index | 2020 | 2021 | 2022 | 2023 | 2024 | 2025 | ||||||||||||||
| Emergent BioSolutions Inc. | $ | 100.00 | $ | 48.52 | $ | 13.18 | $ | 2.68 | $ | 10.67 | $ | 13.79 | ||||||||
| S&P 500 | $ | 100.00 | $ | 128.71 | $ | 105.40 | $ | 133.10 | $ | 166.40 | $ | 196.16 | ||||||||
| Russell 2000 | $ | 100.00 | $ | 114.82 | $ | 91.35 | $ | 106.82 | $ | 119.14 | $ | 134.40 | ||||||||
| S&P SmallCap 600 | $ | 100.00 | $ | 126.82 | $ | 106.40 | $ | 123.48 | $ | 134.22 | $ | 142.30 | ||||||||
| S&P 500 Pharmaceuticals | $ | 100.00 | $ | 125.75 | $ | 136.38 | $ | 136.84 | $ | 148.06 | $ | 188.27 | ||||||||
| S&P 500 Biotechnology | $ | 100.00 | $ | 112.13 | $ | 129.67 | $ | 134.65 | $ | 135.54 | $ | 171.44 | ||||||||
ITEM 6. [RESERVED]
62
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations for each of the two years in the period ended December 31,2025 should be read in conjunction with our consolidated financial statements and accompanying notes and other financial information included elsewhere in this Annual Report on Form 10-K (this “Annual Report”). For a similar discussion and analysis of our results for the year ended December 31, 2024 compared to our results for the year ended December 31, 2023, refer to Part II, Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” of our Annual Report for the year ended December 31, 2024, filed with the United States (“U.S.”) Securities and Exchange Commission (“SEC”) on March 3, 2025. Some of the information contained in this discussion and analysis or set forth elsewhere in this Annual Report includes information with respect to our plans and strategy for our business and financing, as well as forward-looking statements that involve risks and uncertainties. You should carefully review the sections entitled “Risk Factors” and “Cautionary Note Regarding Forward-Looking Statements” in this Annual Report for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
BUSINESS OVERVIEW
Emergent BioSolutions Inc. (“Emergent,” the “Company,” “we,” “us,” and “our”) is a global life sciences company focused on providing innovative preparedness and response solutions addressing accidental, deliberate, and naturally occurring Public Health Threats (“PHTs”). The Company's solutions include a product portfolio, a product development portfolio, and a contract development and manufacturing services (“CDMO”) portfolio.
We are currently focused on the following four PHT categories: chemical, biological, radiological, nuclear and explosives (“CBRNE”); emerging infectious diseases (“EID”); emerging health crises; and acute, emergency and community care. We have a product portfolio of 11 products, 10 of which are owned by the Company, that contribute a substantial portion of our revenue and are sold to government and commercial customers. Additionally, we have a development pipeline consisting of a diversified mix of both pre-clinical and clinical stage product candidates. Finally, we have a fully integrated portfolio of CDMO services which cover development services, drug substance manufacturing and drug product manufacturing and packaging.
The Company structures the business with a focus on markets and customers. As such, the key components of the business structure include the following four product and service categories: Anthrax - Medical Countermeasures (“MCM”) products, Naloxone Commercial products, Smallpox - MCM products and Emergent Bioservices (CDMO) services (“Bioservices”).
The Company manages the business with a focus on three operating segments: (1) a Commercial Products segment consisting of NARCAN® Nasal Spray and KLOXXADO® Nasal Spray, (2) a MCM Products segment consisting of Anthrax - MCM, Smallpox - MCM and Other Products and (3) a Services segment consisting of our Bioservices offerings. Commercial Products and MCM Products are our two reportable segments (see Note 19, “Segment information” in the Notes to Consolidated Financial Statements in Part II, Item 8, of this Form 10-K for more information on our reportable segments).
Commercial Product Segment:
The majority of our Commercial product revenue comes from the following product:
Naloxone Products
•NARCAN® (naloxone HCl) Nasal Spray, an intranasal formulation of naloxone approved by the United States Food and Drug Administration (“FDA”) (including in over-the-counter (“OTC”) form) and Health Canada for the emergency treatment of known or suspected opioid overdose as manifested by respiratory and/or central nervous system depression.
•KLOXXADO® (naloxone HCl) Nasal Spray. In January 2025, the Company announced an agreement with Hikma Pharmaceuticals Inc. (“Hikma”) in which the Company obtained exclusive commercial rights for product sales and marketing in the United States and Canada to Hikma’s KLOXXADO® (naloxone HCl) Nasal Spray, an 8 mg naloxone agent.
MCM Products Segment:
The majority of our MCM product revenue comes from the following products and procured product candidates:
Anthrax - MCM Products
•ANTHRASIL® (Anthrax Immune Globulin Intravenous (human)), the only polyclonal antibody therapeutic licensed by the FDA and Health Canada for the treatment of inhalational anthrax in combination with appropriate antibacterial drugs;
63
•BioThrax® (Anthrax Vaccine Adsorbed), the only vaccine licensed by the FDA for the general use prophylaxis and post-exposure prophylaxis of anthrax disease;
•CYFENDUS® (Anthrax vaccine adsorbed (AVA), adjuvanted), which was approved by the FDA in July 2023 for post-exposure prophylaxis of disease following suspected or confirmed exposure to Bacillus anthracis in persons 18 through 65 years of age when administered in conjunction with recommended antibacterial drugs. CYFENDUS® is procured by certain authorized government buyers for their use; and
•Raxibacumab injection, the first fully human monoclonal antibody therapeutic licensed by the FDA for the treatment and prophylaxis of inhalational anthrax.
Smallpox - MCM Products
•ACAM2000®, (Smallpox (Vaccinia) Vaccine, Live), the only single-dose smallpox vaccine licensed by the FDA for active immunization against smallpox disease for persons determined to be at high risk for smallpox infection;
•CNJ-016® (Vaccinia Immune Globulin Intravenous (Human) (VIGIV)), the only polyclonal antibody therapeutic licensed by the FDA and Health Canada to address certain complications from smallpox vaccination; and
•TEMBEXA®, an oral antiviral formulated as 100 mg tablets and 10 mg/mL oral suspension dosed once weekly for two weeks which has been approved by the FDA for the treatment of smallpox disease caused by variola virus in adult and pediatric patients, including neonates.
Other Products
•BAT® (Botulism Antitoxin Heptavalent (A,B,C,D,E,F,G)-(Equine)), the only heptavalent antitoxin licensed by the FDA and Health Canada for the treatment of symptomatic botulism;
•Ebanga™ (ansuvimab-zykl), a monoclonal antibody with antiviral activity provided through a single IV infusion for the treatment of Ebola. Under the terms of a collaboration with Ridgeback Biotherapeutics (“Ridgeback”), Emergent will be responsible for the manufacturing, sale, and distribution of Ebanga™ in the U.S. and Canada, and Ridgeback will serve as the global access partner for Ebanga™.
Sale of RSDL®
In July 2024, the Company entered into the Stock and Asset Purchase Agreement (the “RSDL® Agreement”) with SERB Pharmaceuticals, through its wholly owned subsidiary BTG International Inc. (collectively, “SERB”), pursuant to which, among other things, the Company sold its worldwide rights to RSDL®, to SERB (the “RSDL® Transaction”). See Note 4, “Divestitures” in the Notes to Consolidated Financial Statements in Part II, Item 8, of this Form 10-K for more information on the sale of RSDL®.
Services Segment:
As of the first quarter of 2025, the Company’s Services operating segment no longer met the quantitative thresholds of a reportable segment and did not meet the aggregation criteria set forth in Accounting Standards Codification 280, Segment Reporting, and as such is categorized within “All other revenues” along with “Contracts and Grants”. See Note 19, “Segment information” in the Notes to Consolidated Financial Statements in Part II, Item 8, of this Form 10-K for more information about the Company’s reportable segments.
64
Other Strategic Activities
May 2024 Organizational Restructuring Plan
In May 2024, the Company initiated an organizational restructuring plan (the “May 2024 Plan”). These strategic actions led to a reduction of the Company’s workforce by approximately 300 employees across all areas of the Company and the elimination of approximately 85 positions that were vacant, as well as the closure of the Company’s Baltimore-Bayview Drug Substance manufacturing facility and Rockville, Maryland Drug Product facility. Decisions regarding the elimination of positions and the closure of manufacturing facilities were subject to local law and consultation requirements in certain countries, as well as the Company’s business needs. The cumulative amount of restructuring charge related to the May 2024 Plan since inception is $18.5 million. All activities related to the May 2024 Plan were substantially completed during the third quarter of 2024. Restructuring costs (benefits) are recognized as an operating expense within the Consolidated Statement of Operations and are classified based on the Company's classification policy for each category of operating expense.
Development milestone payments for CHIKV VLP
In July 2024, Bavarian Nordic announced that the European Medicines Agency had validated the marketing authorization application for CHIKV VLP, which was submitted in June 2024. This approval triggered a milestone payment receivable under the Purchase and Sale Agreement to the Company in the amount of $10.0 million.
In August 2024, Bavarian Nordic announced that the FDA has accepted and granted priority review for the Biologics License Application for CHIKV VLP, which triggered a milestone payment receivable under the Purchase and Sale Agreement to the Company in the amount of $20.0 million.
In February 2025, Bavarian Nordic announced that the FDA approved CHIKV VLP under the Priority Review, which triggered a $30.0 million development milestone payment to the Company. In addition, in February 2025 Bavarian Nordic also announced that the European Commission approved CHIKV VLP, which triggered a $20.0 million development milestone payment to the Company.
August 2024 Organizational Restructuring Plan
In August 2024, the Company initiated an organizational restructuring plan (the “August 2024 Plan”) at the Company’s Lansing facility, which reduced the Company’s workforce by approximately 70 employees, as well as eliminated several open positions. The Company also implemented non-labor optimization efforts, such as reducing the Company’s external and vendor spend. The cumulative amount of restructuring charges related to the August 2024 Plan since inception is $2.5 million. All activities related to the August 2024 Plan were substantially completed during the fourth quarter of 2024. Restructuring costs (benefits) are recognized as an operating expense within the Consolidated Statement of Operations and are classified based on the Company's classification policy for each category of operating expense.
Exclusive commercial rights to KLOXXADO® distribution in U.S. and Canada
In January 2025, the Company announced an agreement with Hikma in which the Company obtained exclusive commercial rights for product sales and marketing in the U.S. and Canada to Hikma’s KLOXXADO® (naloxone HCl) Nasal Spray, an 8 mg naloxone agent.
Securities and shareholder litigation
In September 2024, the Company and the lead plaintiffs in stockholder litigation against the Company entered into an agreement in principle to settle the claims against the Company and each of the Company’s current and former officers and directors. In October 2024, the Court granted preliminary approval of the proposed settlement, ordered notice to the settlement class and the court granted final approval of the settlement in February 2025. Under the settlement, the claims against the Company and its officers and directors were dismissed with prejudice and released in exchange for a payment from the Company of $40.0 million, $30.0 million of which was paid from insurance proceeds, and was funded in the fourth quarter of 2024. The Company recorded the settlement and insurance recoverable amounts as pre-tax operating expense and income, respectively, within “Selling, general and administrative” expenses on the Consolidated Statement of Operations for the year ended December 31, 2024.
65
Sale of Baltimore-Bayview Facility
In March 2025, the Company completed the sale of its Baltimore-Bayview drug substance manufacturing facility to Syngene International (“Syngene”). At closing, Syngene paid a cash purchase price of approximately $36.5 million. Pursuant to the sale, Syngene acquired the assets and equipment associated with the Baltimore-Bayview facility. Emergent retains the rights to secure manufacturing services and capacity at the facility for future growth and pandemic response production in collaboration with Syngene. See Note 4, “Divestitures” in the Notes to Consolidated Financial Statements in Part II, Item 8, of this Form 10-K for further discussion.
August 2025 Settlement
On March 7, 2025, plaintiffs Lincolnshire and Pooja Sayal filed a motion in the United States District Court for the District of Maryland seeking preliminary approval of a stipulation of settlement with regard to the putative shareholder derivative lawsuits filed in 2021 and 2022 (the “Proposed Settlement”). The Proposed Settlement provided that Defendants must cause their insurers to pay to the Company a settlement amount of $15.0 million, less a court-approved fee and expense amount (the “Settlement Amount”). On August 6, 2025, the United States District Court for the District of Maryland granted approval of the Proposed Settlement without modification. The Company received the Settlement Amount in September 2025. Accordingly, during the year ended December 31, 2025, the Company recorded $10.5 million with respect to the Settlement Amount as a reduction of “Selling, general and administrative” expenses on the Consolidated Statement of Operations.
2025 Share Repurchase Program
In March 2025, the Company announced that its Board of Directors had authorized the repurchase of up to $50.0 million of the Company’s common stock (the “2025 Share Repurchase Program”) on or before March 27, 2026. During the year ended December 31, 2025, the Company utilized $25.1 million to repurchase 3.1 million shares at an average price of $8.15 per share, excluding commissions and excise taxes. As of December 31, 2025, the Company had $24.9 million available to repurchase shares under the 2025 Share Repurchase Program. In February 2026, the Company reauthorized the 2025 Share Repurchase Program for the repurchase of up to $50.0 million of the Company's common stock through March 31, 2027.
Senior Unsecured Note Repurchases
In May 2025, the Board authorized the Company to use up to $30.0 million to repurchase Senior Unsecured Notes in open market purchases, privately negotiated transactions or otherwise. During the year ended December 31, 2025, the Company used approximately $8.7 million to repurchase an aggregate principal amount of $10.3 million of the Company’s Senior Unsecured Notes, and the Company recognized a gain on extinguishment of approximately $1.6 million, as recorded in “Gain (loss) on debt extinguishment” on the Consolidated Statement of Operations. As of December 31, 2025, the Company had $19.7 million available to repurchase additional Senior Unsecured Notes.
Term Loan Early Paydown
In the fourth quarter of 2025, the Company executed a partial, early extinguishment of $100.0 million in principal of its $250.0 million term loan with OHA Agency, LLC (the "Term Loan"). The Company recognized a loss on extinguishment of $13.8 million, primarily attributable to the acceleration of unamortized debt issuance costs and prepayment premium, as recorded in “Gain (loss) on debt extinguishment” on the Consolidated Statement of Operations. As of December 31, 2025, the Company had $150.0 million in Term Loan principal remaining. See Note 11, “Debt” in the Notes to Consolidated Financial Statements in Part II, Item 8, of this Form 10-K for more information on the Company's Term Loan Agreement.
66
FINANCIAL OPERATIONS OVERVIEW
Revenues
We generate Commercial Product revenues through the sale of Naloxone products, primarily NARCAN® Nasal Spray, which is sold commercially over-the-counter at retail pharmacies and digital commerce websites as well as through physician-directed or standing order prescriptions at retail pharmacies, health departments, local law enforcement agencies, community-based organizations, substance abuse centers and other federal agencies, as well as KLOXXADO® Nasal Spray, which is currently being integrated into our distribution network, NARCANDirect®. We generate MCM Product revenues from the sale of our marketed products and procured product candidates. The U.S. government (“USG”) is the largest purchaser of our Government - MCM products and primarily purchases our products for the Strategic National Stockpile, a national repository of medical countermeasures including critical antibiotics, vaccines, chemical antidotes, antitoxins, and other critical medical supplies. The USG primarily purchases our products under long-term, firm fixed-price procurement contracts, generally with annual options.
We also generate revenue from our Services segment through our Bioservices portfolio, which is based on our established development and manufacturing infrastructure, technology platforms and expertise. Our services include a fully integrated molecule-to-market Bioservices business offering across development services, drug substance and drug product for small to large pharmaceutical and biotechnology industry and government agencies/non-governmental organizations. From time to time, clients require suite reservations at our various manufacturing sites, which may be considered leases depending on the facts and circumstances.
We have received contracts and grant funding from the USG and other non-governmental organizations to perform R&D activities, particularly related to programs addressing certain CBRNE threats and EIDs.
Our revenue, operating results and profitability vary quarterly based on the timing of production and deliveries, the timing of manufacturing services performed and the nature of our business, which involves providing large scale bundles of products and services as needs arise. We expect continued variability in our quarterly financial results.
Cost of Product Sales and Services
Commercial and MCM Products - The primary expenses that we incur to deliver our Naloxone and MCM products consist of fixed and variable costs. We determine the cost of product sales for products sold during a reporting period based on the average manufacturing cost per unit in the period those units were manufactured. Fixed manufacturing costs include facilities, utilities and amortization of intangible assets. Variable manufacturing costs primarily consist of costs for materials and personnel-related expenses for direct and indirect manufacturing support staff, contract manufacturing operations, sales-based royalties, shipping and logistics. In addition to the fixed and variable manufacturing costs described above, the cost of product sales depends on utilization of available manufacturing capacity. For our commercial sales, other associated expenses include sales-based royalties, shipping, and logistics.
Services - The primary expenses that we incur to deliver our Bioservices offerings consist of fixed and variable costs, including personnel, equipment, and facilities costs. Our manufacturing process includes the production of bulk material and performing drug product work for containment and distribution of biological products. For drug product customers, we receive work in process inventory to be prepared for distribution.
Research and Development (“R&D”) Expenses
We expense R&D costs as incurred. Our R&D expenses consist primarily of:
▪personnel-related expenses;
▪fees to professional service providers for, among other things, analytical testing, independent monitoring or other administration of our clinical trials and obtaining and evaluating data from our clinical trials and non-clinical studies;
▪costs associated with technology transfer and scale up activities throughout the development stage, including internally and through third-party contract manufacturers;
▪costs of Bioservices for our clinical trial material; and
▪costs of materials intended for use and used in clinical trials and R&D.
67
In many cases, we seek funding for development activities from external sources and third parties, such as governments and non-governmental organizations, or through collaborative partnerships. We expect our R&D spending will be dependent upon such factors as the results from our clinical trials, the availability of reimbursement of R&D spending, the number of product candidates under development, the size, structure and duration of any clinical programs that we may initiate, the costs associated with manufacturing and development of our product candidates on a large-scale basis for later stage clinical trials, and our ability to use or rely on data generated by government agencies.
Selling, General and Administrative Expenses
Selling, general and administrative (“SG&A”) expenses consist primarily of personnel-related costs and professional fees in support of our executives, sales and marketing, business development, government affairs, finance, accounting, information technology, legal, human resource functions and other corporate functions. Other costs include facility costs not otherwise included in cost of product sales and Bioservices or R&D expense.
Income taxes
Uncertainty in income taxes is accounted for using a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. We recognize in our financial statements the impact of a tax position if that position is more likely than not of being sustained on audit, based on the technical merits of the position.
Changes in tax laws, rulings, policies, or related legal and regulatory interpretations occur frequently and may have significant favorable or adverse impacts on our effective tax rate. In 2021, the Organization for Economic Cooperation and Development released model rules for a 15% global minimum tax applied to cross-border profits of certain large multinational corporations, known as Pillar Two. Pillar Two has now been enacted by approximately 60 countries, including Ireland. This minimum tax is treated as a period cost beginning in 2024 and its impact is included in the Company's financial results of operations for the current period. The Company is monitoring legislative developments, as well as additional guidance from countries that have enacted legislation.
In July 2025, the One Big Beautiful Bill Act (“OBBBA”) was enacted into law in the United States. The OBBBA includes significant provisions, such as the permanent extension of certain expiring provisions of the Tax Cuts and Jobs Act, modifications to the international tax framework and the restoration of favorable tax treatment for certain business provisions. The legislation has multiple effective dates, with certain provisions effective in 2025 and others implemented through 2027. These impacts did not have a material effect on our tax rate for the year ended December 31, 2025.
Management believes that the assumptions and estimates related to the provision for income taxes are material to the Company’s results of operations.
68
RESULTS OF OPERATIONS
Consolidated and Segment Operating Results:
Year Ended December 31, | |||||||||||||||||||||||
(in millions, except %) | 2025 | 2024 | $ Change | % Change | |||||||||||||||||||
Revenues: | |||||||||||||||||||||||
| Commercial Product sales, net: | |||||||||||||||||||||||
| Naloxone | $ | 226.1 | $ | 398.9 | $ | (172.8) | (43) | % | |||||||||||||||
| Total Commercial Product sales, net | 226.1 | 398.9 | (172.8) | (43) | % | ||||||||||||||||||
| MCM Product sales, net: | |||||||||||||||||||||||
| Anthrax MCM | 114.3 | 138.5 | (24.2) | (17) | % | ||||||||||||||||||
| Smallpox MCM | 266.1 | 277.3 | (11.2) | (4) | % | ||||||||||||||||||
| Other Products | 76.3 | 94.0 | (17.7) | (19) | % | ||||||||||||||||||
| Total MCM Product sales, net | 456.7 | 509.8 | (53.1) | (10) | % | ||||||||||||||||||
All other revenues (1) | 60.1 | 134.9 | (74.8) | (55) | % | ||||||||||||||||||
| Total revenues | $ | 742.9 | $ | 1,043.6 | $ | (300.7) | (29) | % | |||||||||||||||
| Operating expenses: | |||||||||||||||||||||||
Cost of product and services sales, net (2) | 326.2 | 681.3 | (355.1) | (52) | % | ||||||||||||||||||
| Research and development | 53.2 | 70.7 | (17.5) | (25) | % | ||||||||||||||||||
| Selling, general and administrative | 186.1 | 308.0 | (121.9) | (40) | % | ||||||||||||||||||
| Amortization of intangible assets | 65.1 | 65.1 | — | — | % | ||||||||||||||||||
| Impairment of long-lived assets | 12.2 | 27.2 | (15.0) | (55) | % | ||||||||||||||||||
| Total operating expenses | 642.8 | 1,152.3 | (509.5) | (44) | % | ||||||||||||||||||
| Income (loss) from operations | 100.1 | (108.7) | 208.8 | 192 | % | ||||||||||||||||||
| Other income (expense): | |||||||||||||||||||||||
| Interest expense | (59.3) | (71.0) | (11.7) | (16) | % | ||||||||||||||||||
| Gain on sale of business | — | 24.3 | (24.3) | (100) | % | ||||||||||||||||||
| Gain (loss) on debt extinguishment | (12.2) | 0.6 | (12.8) | NM | |||||||||||||||||||
| Other, net | 54.2 | 11.9 | 42.3 | NM | |||||||||||||||||||
| Total other expense, net | (17.3) | (34.2) | (16.9) | (49) | % | ||||||||||||||||||
| Income (loss) before income taxes | 82.8 | (142.9) | 225.7 | 158 | % | ||||||||||||||||||
| Income tax provision | 30.2 | 47.7 | (17.5) | (37) | % | ||||||||||||||||||
| Net income (loss) | $ | 52.6 | $ | (190.6) | $ | 243.2 | 128 | % | |||||||||||||||
(1) "All other revenues" includes Services and Contracts and grants revenue | |||||||||||||||||||||||
(2) Exclusive of intangible assets amortization | |||||||||||||||||||||||
| NM - Not meaningful | |||||||||||||||||||||||
69
Year Ended December 31, 2025 Compared with Year Ended December 31, 2024
Revenues and gross margin
Year Ended December 31, | |||||||||||||||||
(dollars in millions) | 2025 | 2024 | % Change | ||||||||||||||
| Total revenues | $ | 742.9 | $ | 1,043.6 | (29) | % | |||||||||||
| Contracts and grants | 37.7 | 30.0 | 26 | % | |||||||||||||
| Product and services sales, net | $ | 705.2 | $ | 1,013.6 | (30) | % | |||||||||||
| Cost of product and services sales, net | $ | 326.2 | $ | 681.3 | (52) | % | |||||||||||
| Intangible asset amortization | 65.1 | 65.1 | — | % | |||||||||||||
Gross margin (1) | $ | 313.9 | $ | 267.2 | 17 | % | |||||||||||
Gross margin % (1) | 45 | % | 26 | % | |||||||||||||
(1) Gross margin is calculated as product and services sales, net less cost of product and services sales, net and intangible asset amortization. Gross margin percentage is calculated as gross margin divided by product and services sales, net. | |||||||||||||||||
Total revenues decreased $300.7 million, or 29%, to $742.9 million in 2025. The decrease was due to lower Commercial Products revenue of $172.8 million, Services revenue of $82.5 million, and MCM Products revenue of $53.1 million, partially offset by an increase in Contracts and grants revenue of $7.7 million.
Intangible asset amortization remained unchanged at $65.1 million for the years ended December 31, 2025 and 2024.
Gross margin increased $46.7 million, or 17%, to $313.9 million in 2025. Gross margin percentage increased 19 percentage points to 45%. The increase was primarily due to improvements in Services gross margin of $160.5 million and in MCM Products gross margin of $3.2 million, partially offset by a decrease in Commercial Products gross margin of $117.0 million. Gross margin and gross margin percentage excludes Contracts and grants revenues because the related costs are R&D expenses.
See “Reportable Segment Results” for an expanded discussion of revenues and gross profit.
Unallocated corporate operating expenses
R&D Expenses
R&D expenses decreased $17.5 million, or 25%, to $53.2 million in 2025. The decrease was primarily due to write-offs related to program terminations in the second quarter of 2024, coupled with decreases in overhead and severance related costs in the current year. This decrease was partially offset by an increase in costs associated with the Ebanga™ program.
SG&A Expenses
SG&A expenses decreased $121.9 million, or 40%, to $186.1 million in 2025. The decrease was primarily due to a reduction in compensation and other employee related costs as a result of the restructuring initiatives during 2023 and 2024, as well as a decrease in professional services and legal fees related to general corporate initiatives in the prior year. The decline in SG&A was also due to the absence of a one-time expense of $10.0 million recognized in the prior year period and the receipt of a one-time reimbursement of $10.5 million in the current year period related to settlements of our securities and shareholder litigation matters as well as decreases in marketing spend. SG&A expenses as a percentage of total revenue decreased 4 percentage points to 25% for the year ended December 31, 2025.
Impairment of Long-lived Assets
Impairment of long-lived assets decreased $15.0 million, or 55%, to $12.2 million in 2025. The decrease was due to the one time $27.2 million non-cash impairment charge in the second quarter of 2024 related to our Bayview and Rockville asset groups within the Bioservices reporting unit, partially offset by a $12.2 million impairment charge associated with the Maryland warehouse disposal group write-down during 2025.
70
Interest expense
Interest expense decreased $11.7 million, or 16%, to $59.3 million in 2025. The decrease was primarily due to lower interest costs related to our prior syndicated borrowings, partially offset by higher interest expense related to our Term Loan Agreement, and an increase in amortization of debt service costs.
Gain on sale of business
Gain on sale of business decreased $24.3 million, or 100%, due to no sale of business occurring during the year ended December 31, 2025. The gain on sale of business in the prior year was related to the sale of RSDL® to SERB, partially offset by a loss on the sale of the Company’s drug product facility in Baltimore-Camden to Bora.
Gain (loss) on debt extinguishment
Gain (loss) on debt extinguishment decreased $12.8 million to a $12.2 million loss in 2025. The decrease was due to a loss on early extinguishment of $13.8 million for the prepayment of $100.0 million of the Term Loan, primarily attributable to the acceleration of unamortized debt issuance costs and prepayment premium. This was partially offset by a gain on extinguishment of approximately $1.6 million for the principal repurchases of the Company’s Senior Unsecured Notes.
Other, net
Other, net was $54.2 million in income in 2025 compared with $11.9 million in income in 2024. The change of $42.3 million was primarily attributable to a favorable shift from prior-year asset sale losses to asset sale gains in the current year related to our Bayview facility. In addition, the Company recognized $20.0 million of incremental income from milestone payments received in 2025 related to the sale of our travel health business to Bavarian Nordic, as well as favorable impacts from interest income increase and foreign currency, partially offset by a loss from warrant valuation.
Income tax provision
Income tax provision of $47.7 million for the year ended December 31, 2024 decreased $17.5 million to a provision of $30.2 million for the year ended December 31, 2025. The effective tax rate was 36% for the year ended December 31, 2025 as compared to (33)% in 2024. The effective annual tax rate differs from the prior year largely due to jurisdictional mix of income and losses, GILTI, and other permanent items.
71
SEGMENT RESULTS
COMMERCIAL PRODUCTS SEGMENT
Year Ended December 31, | |||||||||||||||||
(dollars in millions) | 2025 | 2024 | % Change | ||||||||||||||
| Revenues | $ | 226.1 | $ | 398.9 | (43) | % | |||||||||||
| Cost of sales | 130.1 | 185.9 | (30) | % | |||||||||||||
| Intangible asset amortization | 37.8 | 37.8 | — | % | |||||||||||||
Gross margin (1) | $ | 58.2 | $ | 175.2 | (67) | % | |||||||||||
Gross margin % (1) | 26 | % | 44 | % | |||||||||||||
| Add back: | |||||||||||||||||
| Intangible asset amortization | 37.8 | 37.8 | — | % | |||||||||||||
| Restructuring costs | 0.2 | — | NM | ||||||||||||||
Segment adjusted gross margin (2) | $ | 96.2 | $ | 213.0 | (55) | % | |||||||||||
Segment adjusted gross margin % (2) | 43 | % | 53 | % | |||||||||||||
(1) Gross margin is calculated as revenues less cost of sales and intangible asset amortization. Gross margin percentage is calculated as gross margin divided by revenues. | |||||||||||||||||
(2) Segment adjusted gross margin, which is a non-GAAP financial measure, for our Commercial Products segment is calculated as gross margin plus intangible asset amortization and restructuring costs. Segment adjusted gross margin percentage, which is a non-GAAP financial measure, is calculated as segment adjusted gross margin divided by segment revenues. The Company’s management utilizes segment adjusted gross margin and segment adjusted gross margin percentage for purposes of evaluating our ongoing operations and for internal planning and forecasting purposes. We believe that these non-GAAP operating measures, when reviewed collectively with our GAAP financial information, provide useful supplemental information to investors in assessing our operating performance. | |||||||||||||||||
NM - Not meaningful | |||||||||||||||||
Year Ended December 31, 2025 Compared with Year Ended December 31, 2024
Naloxone
Naloxone sales decreased $172.8 million, or 43%, to $226.1 million in 2025. The decrease was primarily driven by lower sales of OTC NARCAN® and Canadian sales of branded NARCAN®, driven primarily by increased competition due to generics impacting price and unit sales, partially offset by an increase in KLOXXADO® sales.
Cost of Sales and Gross Margin
Cost of Commercial Products sales decreased $55.8 million, or 30%, to $130.1 million in 2025. The decrease was primarily due to lower sales of OTC NARCAN® and lower Canadian sales of branded NARCAN®, partially offset by an increase in KLOXXADO® sales.
Commercial Products gross margin decreased $117.0 million, or 67%, to $58.2 million in 2025. Commercial Products gross margin percentage decreased 18 percentage points to 26% in 2025. The decrease was primarily due to an unfavorable price and volume mix of OTC NARCAN® and lower Canadian sales of branded NARCAN®, partially offset by an increase in KLOXXADO® sales. Commercial Products segment adjusted gross margin in the current year excludes the impact of intangible asset amortization of $37.8 million and restructuring costs of $0.2 million.
72
MCM PRODUCTS SEGMENT
Year Ended December 31, | |||||||||||||||||
(dollars in millions) | 2025 | 2024 | % Change | ||||||||||||||
| Revenues | $ | 456.7 | $ | 509.8 | (10) | % | |||||||||||
| Cost of sales | 163.1 | 219.4 | (26) | % | |||||||||||||
| Intangible asset amortization | 27.3 | 27.3 | — | % | |||||||||||||
Gross margin (1) | $ | 266.3 | $ | 263.1 | 1 | % | |||||||||||
Gross margin % (1) | 58 | % | 52 | % | |||||||||||||
| Add back: | |||||||||||||||||
| Intangible asset amortization | 27.3 | 27.3 | — | % | |||||||||||||
| Changes in fair value of financial instruments | — | 0.6 | (100) | % | |||||||||||||
Restructuring costs (benefits) | (1.0) | 7.2 | (114) | % | |||||||||||||
| Inventory step-up provision | 5.4 | 6.2 | (13) | % | |||||||||||||
Segment adjusted gross margin (2) | $ | 298.0 | $ | 304.4 | (2) | % | |||||||||||
Segment adjusted gross margin % (2) | 65 | % | 60 | % | |||||||||||||
(1) Gross margin is calculated as revenues less cost of sales and intangible asset amortization. Gross margin percentage is calculated as gross margin divided by revenues. | |||||||||||||||||
(2) Segment adjusted gross margin, which is a non-GAAP financial measure, for our MCM Products segment is calculated as gross margin plus intangible asset amortization, restructuring costs (benefits) and non-cash items related to changes in fair value of financial instruments and inventory step-up provision. Segment adjusted gross margin percentage, which is a non-GAAP financial measure, is calculated as segment adjusted gross margin divided by segment revenues. The Company’s management utilizes segment adjusted gross margin and segment adjusted gross margin percentage for purposes of evaluating our ongoing operations and for internal planning and forecasting purposes. We believe that these non-GAAP operating measures, when reviewed collectively with our GAAP financial information, provide useful supplemental information to investors in assessing our operating performance. | |||||||||||||||||
Year Ended December 31, 2025 Compared with Year Ended December 31, 2024
Anthrax MCM
Anthrax MCM sales decreased $24.2 million, or 17%, to $114.3 million in 2025. The decrease was due to lower volumes of CYFENDUS® sales to the USG and international customers, primarily due to the impact of timing, partially offset by increases in ANTHRASIL® sales to the Canadian government and BioThrax® sales to foreign governments. Anthrax MCM product sales are primarily made under annual purchase options exercised by the USG. Fluctuations in revenues result from the timing of the exercise of annual purchase options, the timing of USG purchases, the availability of governmental funding and Company delivery of orders that follow.
Smallpox MCM
Smallpox MCM sales decreased $11.2 million, or 4%, to $266.1 million in 2025. The decrease was primarily due to lower ACAM2000® sales volumes to both U.S. and non-U.S. customers, partially offset by increases in sales of TEMBEXA® that did not occur in the prior year, and CNJ-016® (VIGIV). Fluctuations in revenues from Smallpox MCM products result from the timing of the exercise of annual purchase options in existing procurement contracts, the timing of USG purchases, the availability of governmental funding and Company delivery of orders that follow.
Other Product Sales
Other Product sales decreased $17.7 million, or 19%, to $76.3 million in 2025. The decrease was primarily due to the sale of RSDL® to SERB during the third quarter of 2024 and a decrease in BAT® foreign sales, partially offset by higher USG BAT® sales due to timing.
73
Cost of Sales and Gross Margin
Cost of MCM Product sales decreased $56.3 million, or 26%, to $163.1 million in 2025. The decrease was primarily due to favorable manufacturing variances mostly due to lower shut-down and severance costs and lower inventory reserves, as well as lower production costs reflecting reduced volumes of ACAM2000® and CYFENDUS®, and no RSDL® related costs in 2025 due to the sale of RSDL® to SERB in the third quarter of 2024. These decreases were partially offset by higher costs for ANTHRASIL® and TEMBEXA® due to higher unit volume.
MCM Products gross margin increased $3.2 million, or 1%, to $266.3 million in 2025. MCM Products gross margin percentage increased 6 percentage points to 58% in 2025. The increase in gross margin percentage was primarily due to a favorable product sales mix which was weighted more heavily towards higher margin products and a decrease in shut-down and severance costs and inventory reserves compared with the prior year period. MCM Product segment adjusted gross margin in the current year excludes the impacts of intangible asset amortization of $27.3 million, inventory step-up provision of $5.4 million, and restructuring benefits of $1.0 million.
ALL OTHER REVENUES
Year Ended December 31, 2025 Compared with Year Ended December 31, 2024
Services Revenues
Services revenues decreased $82.5 million, or 79%, to $22.4 million in 2025. The decrease was primarily attributable to the one time $50.0 million arbitration settlement with Janssen related to the 2022 termination of the Janssen Agreement (the “Settlement Agreement”), which was paid in 2024, coupled with lower revenue from the Company’s Camden facility in the current year, which was sold to Bora in the third quarter of 2024, partially offset by an increase in production at the Company's Winnipeg facility.
Contracts and Grants
Contract and grants revenue increased $7.7 million, or 26%, to $37.7 million in 2025 compared with 2024. The increase was primarily related to work under the EbangaTM program, partially offset by the wind-down of our other funded development initiatives.
74
Financial Condition, Liquidity and Capital Resources
Our financial condition is summarized as follows:
December 31, | |||||||||||||||||
(dollars in millions) | 2025 | 2024 | Change % | ||||||||||||||
| Financial assets: | |||||||||||||||||
| Cash and cash equivalents | $ | 205.4 | $ | 99.5 | 106 | % | |||||||||||
| Restricted cash | 3.7 | 6.1 | (39) | % | |||||||||||||
Total cash, cash equivalents and restricted cash | $ | 209.1 | $ | 105.6 | 98 | % | |||||||||||
| Borrowings: | |||||||||||||||||
| Debt | 572.1 | 663.7 | (14) | % | |||||||||||||
| Total borrowings | $ | 572.1 | $ | 663.7 | (14) | % | |||||||||||
| Working capital: | |||||||||||||||||
| Current assets | $ | 662.5 | $ | 598.7 | 11 | % | |||||||||||
| Current liabilities | 132.2 | 162.4 | (19) | % | |||||||||||||
| Total working capital | $ | 530.3 | $ | 436.3 | 22 | % | |||||||||||
| NM - Not Meaningful | |||||||||||||||||
Principal Sources of Capital Resources
We have historically financed our operating and capital expenditures through existing cash and cash equivalents, cash from operations, development contracts and grant funding and borrowings under various credit agreements, including our Term Loan Agreement and other lines of credit we have established from time to time. We also occasionally obtain financing from the sale of our common stock upon exercise of stock options. As of December 31, 2025, we had unrestricted cash and cash equivalents of $205.4 million and available borrowing capacity of up to $100.0 million under the Revolving Credit Agreement. As of December 31, 2025, the Company believes that its sources of liquidity, including debt and cash flows from operating activities, are adequate to fund its operations for at least the next twelve months from the issuance of these consolidated financial statements.
2025 Share Repurchase Program
In March 2025, the Company announced that its Board of Directors had authorized the repurchase of up to $50.0 million of the Company’s common stock (the “2025 Share Repurchase Program”) on or before March 27, 2026. In February 2026, the Company reauthorized the 2025 Share Repurchase Program for the repurchase of up to $50.0 million of the Company's common stock through March 31, 2027. Repurchases under the 2025 Share Repurchase Program may be made from time to time on the open market or in privately negotiated transactions. The timing and amount of any shares repurchased will be determined by the Company’s management based on its evaluation of market conditions and other factors, including the market price of the Company’s common stock, macroeconomic environment and other investment opportunities consistent with applicable law. The 2025 Share Repurchase Program may be suspended or discontinued at any time. The Inflation Reduction Act of 2022, imposed a nondeductible 1% excise tax on the net value of certain stock repurchases made after December 31, 2022. Excise tax accrued during the year ended December 31, 2025 was $0.3 million.
During the year ended December 31, 2025, the Company utilized $25.1 million to repurchase 3.1 million shares at an average price of $8.15 per share, excluding commissions and excise taxes. As of December 31, 2025, the Company had $24.9 million available to repurchase shares under the 2025 Share Repurchase Program.
Senior Unsecured Note Repurchase
In May 2025, the Board of Directors authorized the Company to repurchase up to $30.0 million aggregate principal amount of the Company’s Senior Unsecured Notes. The Company may seek to opportunistically use this authority to repurchase its Senior Unsecured Notes in open market purchases, privately negotiated transactions or otherwise. Any such repurchases will depend upon prevailing market conditions, our liquidity requirements, contractual restrictions, applicable securities law and other factors. During the year ended December 31, 2025, we repurchased $10.3 million aggregate principal amount of the Company’s Senior Unsecured Notes for $8.7 million in cash, including fees, and recognized a gain on extinguishment of approximately $1.6 million. As of December 31, 2025, we had $19.7 million available to repurchase additional Senior Unsecured Notes.
75
Cash Flows
The following table provides information regarding our cash flows for the years ended December 31, 2025 and 2024.
Year Ended December 31, | |||||||||||
(dollars in millions) | 2025 | 2024 | |||||||||
| Net cash provided by (used in): | |||||||||||
| Operating activities | $ | 170.6 | $ | 58.7 | |||||||
| Investing activities | 69.4 | 125.2 | |||||||||
| Financing activities | (136.6) | (190.0) | |||||||||
| Effect of exchange rate changes on cash, cash equivalents and restricted cash | 0.1 | — | |||||||||
| Net change in cash, cash equivalents and restricted cash | $ | 103.5 | $ | (6.1) | |||||||
Operating Activities:
Net cash provided by operating activities increased $111.9 million in 2025 compared with 2024. The increase in net cash provided by operating activities was primarily due to higher net income excluding non-cash items due to improved operating performance in 2025, partially offset by unfavorable changes in working capital, including a significant non-cash write-off in 2024 related to the Janssen Agreement termination included in prepaid expenses and other assets, and higher cash payments related to accrued expenses and income taxes.
Investing Activities:
Net cash provided by investing activities decreased $55.8 million in 2025 compared with 2024. The decrease in net cash provided by investing activities was primarily attributable to the significant cash inflows in 2024 related to the sale of RSDL® and the sale of the Baltimore-Camden facility. The decrease was partially offset by greater aggregate milestone payments received in connection with the sale of the Company’s travel health business to Bavarian Nordic in 2025, proceeds from the sale of property, plant and equipment, including the sale of our Baltimore-Bayview facility to Syngene, and a reduction in capital expenditures compared with the prior year.
Financing Activities:
Net cash used in financing activities decreased $53.4 million in 2025 compared with 2024. The decrease in cash used in financing activities was driven by significant net financing outflows in 2024 related to the debt refinancing, primarily due to the principal repayments on the Company’s former revolving and term loan facilities and associated debt issuance costs. The 2025 financing outflows included the recent principal repayment on the Company's term loan facility and repurchases under the Company’s 2025 Share Repurchase Program and Senior Unsecured Note repurchases, which partially offset the year-over-year decrease.
Debt
As of December 31, 2025, the Company has $589.7 million of fixed and variable rate debt with varying maturities, with no payable amounts within 12 months (see Note 11, “Debt” in the Notes to Consolidated Financial Statements in Part II, Item 8. of this Form 10-K).
Uncertainties and Trends Affecting Funding Requirements
We expect to continue to fund our short-term and long-term anticipated operating expenses, capital expenditures and debt service requirements, any future debt repurchases and any future repurchases of our common stock from the following sources:
•existing cash and cash equivalents;
•net proceeds from the sale of our products and Bioservices;
•development contracts and grant funding;
•proceeds from potential asset sales; and
•our Term Loan Agreement and Revolving Loans.
76
There are numerous risks and uncertainties associated with product sales and with the development and commercialization of our product candidates. We may seek additional external financing to provide additional financial flexibility. Our future capital requirements will depend on many factors, including (but not limited to):
•the level, timing and cost of product sales and services sales;
•the extent to which we acquire or invest in and integrate companies, businesses, products or technologies;
•the acquisition of new facilities and capital improvements to new or existing facilities;
•the payment obligations under our indebtedness;
•the scope, progress, results and costs of our development activities;
•our ability to obtain funding from collaborative partners, government entities and non-governmental organizations for our development programs; and
•the costs of commercialization activities, including product marketing, sales and distribution.
If our capital resources are insufficient to meet our future capital requirements, we will need to finance our cash needs through public or private equity or debt offerings, bank loans, collaboration and licensing arrangements, cost reductions, assets sales or a combination of these options.
If we raise funds by issuing equity securities, our stockholders may experience dilution. Public or bank debt financing, if available, may involve agreements that include covenants, like those contained in our Senior Unsecured Notes, our Term Loan Agreement and our Revolving Credit Agreement, which could limit or restrict our ability to take specific actions, such as incurring additional debt, making capital expenditures, pursuing acquisition opportunities, buying back shares or declaring dividends. If we raise funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish valuable rights to our technologies or product candidates or grant licenses on terms that may not be favorable to us.
Economic conditions, including market volatility and adverse impacts on financial markets, may make it more difficult to obtain financing on attractive terms, or at all. Any new debt funding, if available, may be on terms less favorable to us than our Senior Unsecured Notes, our Term Loan Agreement or our Revolving Credit Agreement. If financing is unavailable or lost, our business, operating results, financial condition and cash flows would be adversely affected, and we could be forced to delay, reduce the scope of or eliminate many of our planned activities.
Unused Credit Capacity
Available room under the commitments with respect to the Revolving Credit Agreement (the “Revolving Loans") as of December 31, 2025 and 2024 was:
December 31, | |||||||||||
(in millions) | 2025 | 2024 | |||||||||
| Total Capacity | $ | 100.0 | $ | 100.0 | |||||||
| Unused Capacity | $ | 100.0 | $ | 100.0 | |||||||
Contractual Obligations
As of December 31, 2025, the Company has contractual obligations related to lease arrangements and purchase commitments. The lease arrangements are for certain equipment and facilities. As of December 31, 2025, the Company had fixed lease payment obligations of $16.9 million, with $3.4 million due within 12 months. The Company has non-cancelable purchase commitments of $474.8 million, with an estimated $142.9 million being due within 12 months.
77
Critical Accounting Policies and Estimates
Our consolidated financial statements and related disclosures are prepared in accordance with U.S. GAAP, which requires management to make estimates, judgments and assumptions that affect the amounts reported. Note 2, “Summary of significant accounting policies” of the Notes to Consolidated Financial Statements in Part II, Item 8. of this Form 10-K describes the accounting policies and methods used in the preparation of the Company’s consolidated financial statements. Management considers an accounting policy to be critical if it is important to reporting our financial condition and results of operations, and if it requires significant judgment and estimates on the part of management in its application. Management bases its estimates on historical experience and on various other assumptions it believes to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities and the reported amounts of revenues and expenses that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Management believes the Company’s critical accounting policies and estimates are those related to revenue recognition, long-lived assets and inventories, net.
Revenue Recognition
The Company's product sales are recognized at a point-in-time generally upon delivery to the customer, depending on the performance obligation which the Company is delivering. Certain of the Company's development contracts and grants arrangements which are cost-plus-fee contracts, and Bioservices arrangements, are generally recognized on a percentage of completion basis utilizing a cost-to-cost method or input method, respectively. Revenues are recognized as a percentage of the work completed during the period in an amount that reflects the percentage of the consideration which the Company expects to receive in exchange for the product or services. Estimating costs is subjective and requires assumptions about future activity and cost drivers.
For contracts with multiple performance obligations, the Company allocates the contract price to each performance obligation on a relative standalone selling price basis using the Company’s best estimate of the standalone selling price of each distinct product or service in the contract. Certain contracts may include lease components which are recognized under Accounting Standards Codification ("ASC") 842. The primary method used to estimate standalone selling price is the price observed in standalone sales to customers, however when prices in standalone sales are not available the Company may use third-party pricing for similar products or services or estimate the standalone selling price based on the best available information.
Revenues for Naloxone products are recorded at the net sales price (transaction price), which includes estimates of variable consideration for which reserves are established. Estimates of variable consideration include allowance for returns, specialty distributor fees, wholesaler fees and prompt payment discounts. Revenues from OTC NARCAN® are recognized to the extent that it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with such variable consideration is subsequently resolved. The Company considers several factors in the estimation process for the allowance for returns of OTC NARCAN®, including inventory levels within the distribution channel, product shelf life and historical return activity, including activity for product sold for which the return period has passed, as well as other relevant factors. Because returned product cannot be resold, there is no corresponding asset for product returns.
Revenue recognition is material to our business most notably because the nature of our product sales and development contracts requires us to apply significant judgment in estimating costs, allocating transaction prices to multiple performance obligations, and assessing variable consideration. Because these judgments are inherently subjective, changes in underlying assumptions could materially affect the amount and timing of revenue recognized. For additional information on our revenues, refer to Note 14, "Revenue recognition" in the Notes to Consolidated Financial Statements in Part II, Item 8. of this Form 10-K.
78
Long-lived assets
Long-lived assets such as finite lived intangible assets and property, plant and equipment are not required to be tested for impairment annually, instead they are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. These events or changes in circumstances may include a significant deterioration of operating results, changes in business plans or changes in anticipated future cash flows. If an impairment indicator is present, we evaluate recoverability of assets to be held-and-used by a comparison of the carrying value of the assets with future undiscounted net cash flows expected to be generated by the assets. We group assets at the lowest level for which there are identifiable cash flows that are largely independent of the cash flows generated by other asset groups. If the total of the expected undiscounted future cash flows is less than the carrying amount of the asset group, we estimate the fair value of the asset group to determine whether an impairment loss should be recognized. Impairment would then be measured as the excess of the asset’s carrying value over its fair value. Fair value is typically determined by discounting the future cash flows associated with that asset. Significant judgments used for long-lived asset impairment assessments include identifying the appropriate asset groupings and primary assets within those groupings, determining whether events or circumstances indicate that the carrying amount of the asset may not be recoverable, determining the future cash flows for the assets involved and assumptions applied in determining fair value, which include, reasonable discount rates, growth rates, market risk premiums and other assumptions about the economic environment.
Inventories, net
Inventories are stated at the lower of cost or net realizable value with cost being determined using a standard cost method, which approximates actual cost. Actual cost consists primarily of material, labor and manufacturing overhead expenses (including fixed production-overhead costs) and includes the services and products of third-party suppliers. For internally manufactured inventory, the Company determines normal capacity for each production facility and allocates fixed production-overhead costs on that basis. The Company records inventory acquired in business combinations utilizing the comparative sales method, which estimates the expected sales price reduced for all costs expected to be incurred to complete/dispose of the inventory with a profit on those costs. The Company analyzes its inventory each reporting period and records a reserve to reduce the cost basis to net realizable value for inventory that has become obsolete, expired, slow-moving or short-dated based on customer requirements. Reserves for excess and obsolete inventory are relieved when the related inventory is disposed of through scrap or sale.
Significant New Accounting Pronouncements
See Note 2, “Summary of significant accounting policies” in the Notes to Consolidated Financial Statements in Part II, Item 8. of this Form 10-K.
79
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
For a discussion of additional risks arising from our operations, see “Item 1A—Business—Risk Factors” in this 2025 Annual Report.
Market Risks
We have interest rate and foreign currency market risk. Because of the short-term maturities of our cash and cash equivalents, we believe that an increase in market rates would likely not have a significant impact on the realized value of our investments.
Interest Rate Risk
We have debt with a mix of fixed and variable rates of interest and we are satisfied with the current fix-float mix of the Company's debt portfolio. Floating rate debt carries interest based generally on the secured overnight financing (“SOFR”), as defined in our Term Loan Agreement, plus an applicable margin. Increases in interest rates could result in an increase in interest payments for our floating rate debt. We maintain an undrawn Revolver Loan that, if utilized, would bear interest and therefore could expose us to interest rate risk. See Note 11, "Debt" in the Notes to Consolidated Financial Statements in Part II, Item 8. of this Form 10-K.
From time to time, we may use derivative instruments to manage our interest rate risk and market risk exposure.
We have assessed our exposure to changes in interest rates by analyzing the sensitivity to our operating results assuming various changes in market interest rates. A hypothetical increase of one percentage point in the SOFR rate as of December 31, 2025 would increase our interest expense by approximately $1.5 million annually.
Foreign Currency Exchange Rate Risk
We have exposure to foreign currency exchange rate fluctuations worldwide and primarily with respect to the Euro, Canadian dollar, Singapore dollar, Swiss franc, and British pound. We manage our foreign currency exchange rate risk primarily by either entering into foreign currency hedging transactions or incurring operating expenses in the local currency in the countries in which we operate, to the extent practicable. We currently do not hedge all of our foreign currency exchange exposure and the movement of foreign currency exchange rates could have an adverse or positive impact on our results of operations.
80
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Emergent BioSolutions Inc. and Subsidiaries
| Index to Consolidated Financial Statements | Page | ||||
81
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Emergent BioSolutions Inc.
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Emergent BioSolutions Inc. and subsidiaries (the Company) as of December 31, 2025 and 2024, the related consolidated statements of operations, comprehensive income (loss), changes in stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2025, and the related notes and financial statement schedule listed in the Index at Item 15 (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2025 and 2024, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2025, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2025, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework), and our report dated February 26, 2026 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the account or disclosure to which it relates.
82
Reserve for Excess and Obsolete Finished Goods Inventories | ||||||||
| Description of the Matter | The Company had total inventories, net of $ Auditing the Company’s assessment for finished goods inventory reserves was subjective and involved auditor judgment as the estimate relies on factors that are outside of the Company’s control. In particular, the analysis is dependent on assumptions such as the expected timing of delivery for customer orders and the shelf-life requirements of customers. | |||||||
| How We Addressed the Matter in Our Audit | We obtained an understanding, evaluated the design, and tested the operating effectiveness of the Company's control over the finished goods inventory reserve process, which included management’s review of the assumptions, and the completeness and accuracy of the data underlying the finished goods inventory reserve analysis. Our audit procedures included, among others, assessing the appropriateness of the Company’s methodology and the significant assumptions used in the analysis for finished goods inventory reserves. We tested the mathematical accuracy of the calculations and audited the completeness and accuracy of the underlying inputs used, such as expiration dates and customer shelf-life requirements. In addition, we performed inquiries of management and inspected documentation, such as customer contracts, to evaluate the reasonableness of the Company’s estimate. | |||||||
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2004.
February 26, 2026
83
Emergent BioSolutions Inc. and Subsidiaries
Consolidated Balance Sheets
(in millions, except per share data)
December 31, | |||||||||||
2025 | 2024 | ||||||||||
| ASSETS | |||||||||||
| Current assets: | |||||||||||
| Cash and cash equivalents | $ | $ | |||||||||
| Restricted cash | |||||||||||
| Accounts receivable, net | |||||||||||
| Inventories, net | |||||||||||
| Prepaid expenses and other current assets | |||||||||||
| Total current assets | |||||||||||
| Property, plant and equipment, net | |||||||||||
| Intangible assets, net | |||||||||||
| Other assets | |||||||||||
| Total assets | $ | $ | |||||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | |||||||||||
| Current liabilities: | |||||||||||
| Accounts payable | $ | $ | |||||||||
| Accrued expenses | |||||||||||
| Accrued compensation | |||||||||||
| Current tax liability | |||||||||||
| Other current liabilities | |||||||||||
| Total current liabilities | |||||||||||
| Debt | |||||||||||
| Deferred tax liability | |||||||||||
| Other liabilities | |||||||||||
| Total liabilities | |||||||||||
| Stockholders’ equity: | |||||||||||
Preferred stock, $ | |||||||||||
Common stock, $ | |||||||||||
Treasury stock, at cost, | ( | ( | |||||||||
| Additional paid-in capital | |||||||||||
| Accumulated other comprehensive loss, net | ( | ( | |||||||||
| Accumulated deficit | ( | ( | |||||||||
| Total stockholders’ equity | |||||||||||
| Total liabilities and stockholders’ equity | $ | $ | |||||||||
The accompanying notes are an integral part of the consolidated financial statements.
84
Emergent BioSolutions Inc. and Subsidiaries
Consolidated Statements of Operations
(in millions, except per share data)
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Revenues: | |||||||||||||||||
| Product and services sales, net | $ | $ | $ | ||||||||||||||
| Contracts and grants | |||||||||||||||||
| Total revenues | |||||||||||||||||
| Operating expenses: | |||||||||||||||||
Cost of product and services sales, net (1) | |||||||||||||||||
| Research and development | |||||||||||||||||
| Selling, general and administrative | |||||||||||||||||
| Amortization of intangible assets | |||||||||||||||||
| Goodwill impairment | |||||||||||||||||
| Impairment of long-lived assets | |||||||||||||||||
| Total operating expenses | |||||||||||||||||
| Income (loss) from operations | ( | ( | |||||||||||||||
| Other income (expense): | |||||||||||||||||
| Interest expense | ( | ( | ( | ||||||||||||||
| Gain on sale of business | |||||||||||||||||
| Gain (loss) on debt extinguishment | ( | ||||||||||||||||
| Other, net | |||||||||||||||||
| Total other income (expense), net | ( | ( | ( | ||||||||||||||
| Income (loss) before income taxes | ( | ( | |||||||||||||||
| Income tax provision | |||||||||||||||||
| Net income (loss) | $ | $ | ( | $ | ( | ||||||||||||
| Earnings (loss) per common share | |||||||||||||||||
| Basic | $ | $ | ( | $ | ( | ||||||||||||
| Diluted | $ | $ | ( | $ | ( | ||||||||||||
| Weighted average shares outstanding | |||||||||||||||||
| Basic | |||||||||||||||||
| Diluted | |||||||||||||||||
(1) Exclusive of intangible assets amortization | |||||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
85
Emergent BioSolutions Inc. and Subsidiaries
Consolidated Statements of Comprehensive Income (Loss)
(in millions)
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Net income (loss) | $ | $ | ( | $ | ( | ||||||||||||
| Other comprehensive income (loss), net of tax: | |||||||||||||||||
| Foreign currency translation adjustments, net | ( | ||||||||||||||||
| Unrealized gains on hedging activities | |||||||||||||||||
| Reclassification adjustment for gains on hedging activities | ( | ||||||||||||||||
| Reclassification adjustment for gains on pension benefit obligation | ( | ||||||||||||||||
| Total other comprehensive income (loss), net of tax | ( | ( | |||||||||||||||
| Comprehensive income (loss), net of tax | $ | $ | ( | $ | ( | ||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
86
Emergent BioSolutions Inc. and Subsidiaries
Consolidated Statements of Cash Flows
(in millions)
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Operating Activities | |||||||||||||||||
| Net income (loss) | $ | $ | ( | $ | ( | ||||||||||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | |||||||||||||||||
| Share-based compensation expense | |||||||||||||||||
| Depreciation and amortization | |||||||||||||||||
| Change in fair value of contingent obligations, net | |||||||||||||||||
| Amortization of deferred financing costs | |||||||||||||||||
| Deferred income taxes | ( | ( | ( | ||||||||||||||
| Noncash gain on sale of business | ( | ( | |||||||||||||||
| Change in fair value of warrant liability | |||||||||||||||||
| Goodwill impairment | |||||||||||||||||
| Impairment of long-lived assets | |||||||||||||||||
| Loss on disposal of assets | |||||||||||||||||
| Other | ( | ||||||||||||||||
| Changes in operating assets and liabilities: | |||||||||||||||||
| Accounts receivable | ( | ( | |||||||||||||||
| Inventories | ( | ( | |||||||||||||||
| Prepaid expenses and other assets | |||||||||||||||||
| Accounts payable | ( | ( | |||||||||||||||
| Accrued expenses and other liabilities | ( | ( | |||||||||||||||
| Long-term incentive plan accrual | |||||||||||||||||
| Accrued compensation | ( | ( | ( | ||||||||||||||
| Income taxes receivable and payable, net | ( | ( | |||||||||||||||
| Contract liabilities | ( | ||||||||||||||||
| Net cash provided by (used in) operating activities | ( | ||||||||||||||||
| Investing Activities | |||||||||||||||||
| Purchases of property, plant and equipment | ( | ( | ( | ||||||||||||||
| Proceeds from sale of property, plant and equipment | |||||||||||||||||
| Milestone payments from prior asset divestiture | ( | ||||||||||||||||
| Purchase of convertible note receivable | ( | ||||||||||||||||
| Proceeds from sale of business | |||||||||||||||||
| Net cash provided by investing activities | |||||||||||||||||
| Financing Activities | |||||||||||||||||
| Proceeds from the issuance of debt, net of lender fees | |||||||||||||||||
| Proceeds allocated to warrants issued in conjunction with debt | |||||||||||||||||
| Proceeds allocated to common stock issued in conjunction with debt | |||||||||||||||||
| Principal payments on term loan facility | ( | ( | ( | ||||||||||||||
| Proceeds from revolving credit facility | |||||||||||||||||
| Principal payments on revolving credit facility | ( | ( | |||||||||||||||
| Debt issuance costs | ( | ||||||||||||||||
| Proceeds from issuance of common stock upon exercise of stock options | |||||||||||||||||
| Repurchase of debt | ( | ||||||||||||||||
| Prepayment premium on debt principal payment | ( | ||||||||||||||||
| Purchases of treasury stock | ( | ||||||||||||||||
| Proceeds from share-based compensation activity | |||||||||||||||||
| Proceeds from at-the-market sale of stock, net of commissions and expenses | |||||||||||||||||
| Taxes paid for share-based compensation activity | ( | ( | ( | ||||||||||||||
| Net cash used in financing activities: | ( | ( | ( | ||||||||||||||
| Effect of exchange rate changes on cash, cash equivalents and restricted cash | ( | ||||||||||||||||
| Net change in cash, cash equivalents and restricted cash | ( | ( | |||||||||||||||
| Cash, cash equivalents and restricted cash, beginning of period | |||||||||||||||||
| Cash, cash equivalents and restricted cash, end of period | $ | $ | $ | ||||||||||||||
| Supplemental cash flow disclosures: | |||||||||||||||||
| Cash paid for interest | $ | $ | $ | ||||||||||||||
| Cash paid for income taxes, net of refunds | $ | $ | $ | ||||||||||||||
| Non-cash investing and financing activities: | |||||||||||||||||
| Purchases of property, plant and equipment unpaid at period end | $ | $ | $ | ||||||||||||||
| Gain (loss) on extinguishments of debt | $ | ( | $ | $ | |||||||||||||
| Issuance of common stock in conjunction with debt | $ | $ | $ | ||||||||||||||
| Excise tax liability accrued for common stock repurchases | $ | $ | $ | ||||||||||||||
| Reconciliation of cash and cash equivalents and restricted cash: | |||||||||||||||||
| Cash and cash equivalents | $ | $ | $ | ||||||||||||||
| Restricted cash | |||||||||||||||||
| Total | $ | $ | $ | ||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
87
Emergent BioSolutions Inc. and Subsidiaries
Consolidated Statement of Changes in Stockholders' Equity
(in millions, except per share data)
$ Common Stock | Additional Paid-In Capital | Treasury Stock | Accumulated Other Comprehensive Income (Loss) | Retained Earnings (Accumulated Deficit) | Total Stockholders' Equity | |||||||||||||||||||||||||||||||||||||||||||||
Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2022 | $ | $ | ( | $ | ( | $ | $ | $ | ||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | ( | ( | ||||||||||||||||||||||||||||||||||||||||||
| Share-based compensation activity | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||
| At-the-market sale of stock, net of commissions and expenses | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||
| Other comprehensive loss, net of tax | — | — | — | — | — | ( | — | ( | ||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2023 | $ | $ | ( | $ | ( | $ | ( | $ | ( | $ | ||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | ( | ( | ||||||||||||||||||||||||||||||||||||||||||
| Share-based compensation activity | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||
| Other comprehensive income, net of tax | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2024 | $ | $ | ( | $ | ( | $ | ( | $ | ( | $ | ||||||||||||||||||||||||||||||||||||||||
| Net income | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||
| Share-based compensation activity | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||
| Repurchase of common stock | — | — | — | ( | ( | — | — | ( | ||||||||||||||||||||||||||||||||||||||||||
| Other comprehensive loss, net of tax | — | — | — | — | — | ( | — | ( | ||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2025 | $ | $ | ( | $ | ( | $ | ( | $ | ( | $ | ||||||||||||||||||||||||||||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
88
Emergent BioSolutions Inc. and Subsidiaries
Notes to Consolidated Financial Statements
(dollar and share amounts in tables expressed in millions, except per share data)
1. Nature of the business and organization
Organization and business
Emergent BioSolutions Inc. (“Emergent,” the “Company,” “we,” “us,” and “our”) is a global life sciences company focused on providing innovative preparedness and response solutions addressing accidental, deliberate, and naturally occurring Public Health Threats (“PHTs”). The Company's solutions include a product portfolio, a product development portfolio, and a contract development and manufacturing (“CDMO”) services portfolio.
The Company is focused on the following four PHT categories: chemical, biological, radiological, nuclear and explosives (“CBRNE”); emerging infectious diseases (“EID”); emerging health crises; and acute, emergency and community care. As of December 31, 2025, the Company has a product portfolio of 11 products consisting of vaccines, therapeutics, and drug-device combination products, including KLOXXADO® Nasal Spray for which the Company obtained certain exclusive commercial rights for product sales and marketing in the United States and Canada. The revenue generated by the products comprises a substantial portion of the Company's revenue. The Company structures the business with a focus on markets and customers. As such, the key components of the business structure include the following four product and service categories: Anthrax - Medical Countermeasures (“MCM”) products, Naloxone Commercial products, Smallpox - MCM products and Emergent Bioservices (CDMO) (“Bioservices”).
The Company manages the business with a focus on three operating segments: (1) a Commercial Products segment consisting of NARCAN® Nasal Spray and KLOXXADO® Nasal Spray, as described below; (2) a MCM Products segment consisting of our Anthrax - MCM, Smallpox - MCM and Other Products, described below and (3) a Services segment consisting of our Bioservices offerings. Commercial Products and MCM Products are our two reportable segments (see Note 19, “Segment information” for more information on our reportable segments).
The Company's products and services include:
Commercial Products Segment:
Naloxone Products
•NARCAN® (naloxone HCl) Nasal Spray is an intranasal formulation of naloxone approved by the United States Food and Drug Administration (“FDA”) (including in over-the-counter (“OTC”) form) and Health Canada for the emergency treatment of known or suspected opioid overdose as manifested by respiratory and/or central nervous system depression.
•KLOXXADO® (naloxone HCl) Nasal Spray. In January 2025, the Company announced an agreement with Hikma Pharmaceuticals Inc. (“Hikma”) in which the Company obtained exclusive commercial rights for product sales and marketing in the United States and Canada to Hikma’s KLOXXADO® (naloxone HCl) Nasal Spray, an 8 mg naloxone agent.
MCM Products Segment:
Anthrax - MCM Products
•ANTHRASIL® (Anthrax Immune Globulin Intravenous (human)), the only polyclonal antibody therapeutic licensed by the FDA and Health Canada for the treatment of inhalational anthrax in combination with appropriate antibacterial drugs;;
•BioThrax® (Anthrax Vaccine Adsorbed), the only vaccine licensed by the FDA for the general use prophylaxis and post-exposure prophylaxis of anthrax disease;
•CYFENDUS® (Anthrax vaccine adsorbed (AVA), adjuvanted) which was approved by the FDA in July 2023 for post-exposure prophylaxis of disease following suspected or confirmed exposure to Bacillus anthracis in persons 18 through 65 years of age when administered in conjunction with recommended antibacterial drugs. CYFENDUS® is procured by certain authorized government buyers for their use; and
•Raxibacumab injection, the first fully human monoclonal antibody therapeutic licensed by the FDA for the treatment and prophylaxis of inhalational anthrax.
89
Smallpox - MCM Products
•ACAM2000®, (Smallpox (Vaccinia) Vaccine, Live), the only single-dose smallpox vaccine licensed by the FDA for active immunization against smallpox disease for persons determined to be at high risk for smallpox infection;
•CNJ-016® (Vaccinia Immune Globulin Intravenous (Human) (VIGIV)), the only polyclonal antibody therapeutic licensed by the FDA and Health Canada to address certain complications from smallpox vaccination; and
•TEMBEXA®, an oral antiviral formulated as 100 mg tablets and 10 mg/mL oral suspension dosed once weekly for two weeks which has been approved by the FDA for the treatment of smallpox disease caused by variola virus in adult and pediatric patients, including neonates.
Other Products
•BAT® (Botulism Antitoxin Heptavalent (A,B,C,D,E,F,G)-(Equine)), the only heptavalent antitoxin licensed by the FDA and Health Canada for the treatment of symptomatic botulism; and
•Ebanga™ (ansuvimab-zykl), a monoclonal antibody with antiviral activity provided through a single IV infusion for the treatment of Ebola. Under the terms of a collaboration with Ridgeback Biotherapeutics (“Ridgeback”), Emergent will be responsible for the manufacturing, sale, and distribution of Ebanga™ in the U.S. and Canada, and Ridgeback will serve as the global access partner for Ebanga™.
Sale of RSDL®
In July 2024, the Company entered into the Stock and Asset Purchase Agreement (the “RSDL® Agreement”) with SERB Pharmaceuticals, through its wholly owned subsidiary BTG International Inc. (collectively, “SERB”), pursuant to which, among other things, the Company sold its worldwide rights to RSDL® to SERB (the “RSDL® Transaction”). See Note 4, “Divestitures” for more information on the RSDL® Transaction.
Services Segment:
90
2. Summary of significant accounting policies
Basis of presentation and consolidation
Our financial statements are prepared in conformity with U.S. generally accepted accounting principles (“GAAP”). The accompanying consolidated financial statements include the accounts of Emergent and its wholly owned subsidiaries. All significant inter-company accounts and transactions have been eliminated in consolidation. For presentation purposes, reclassifications of certain items have been made for comparability.
Use of estimates
The preparation of financial statements requires management to make estimates, judgments and assumptions that affect reported amounts and disclosures in the consolidated financial statements and accompanying notes. Due to the inherent uncertainty involved in making those estimates, judgments and assumptions, actual results could differ from those estimates. Our most significant estimates relate to revenue recognitions and the assessment of the recoverability of definite lived intangible assets and other long-lived assets. Other estimates include allowances for expected credit losses, inventory, depreciation and amortization, contingent consideration, share-based compensation, warrants, income taxes, and other contingencies. Management continually re-evaluates its estimates, judgments and assumptions and basis its estimates on historical trends, projections, current experience and other assumptions that it believes are reasonable. These estimates are sometimes complex, sensitive to changes in assumptions and require fair value determinations using Level 3 fair value measurements.
Cash and cash equivalents and restricted cash
Cash equivalents are highly liquid investments with a maturity of 90 days or less at the date of purchase and consist of time deposits and investments in money market funds with commercial banks and financial institutions. Also, the Company maintains cash balances with financial institutions in excess of insured limits. Restricted cash includes cash that is not readily available for use in the Company's operating activities. Restricted cash is primarily comprised of cash pledged under letters of credit.
Warrants
The Company accounts for Warrants as either equity instruments, liabilities or derivative liabilities in accordance with ASC 480, Distinguishing Liabilities from Equity (“ASC 480”) and/or ASC 815, Derivatives and Hedging (“ASC 815”), depending on the specific terms of the applicable warrant agreement.
Fair value measurements
Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability, an exit price, in the principal or most advantageous market for the asset or liability in an orderly transaction between market participants on the measurement date. Valuation techniques used to measure fair value must maximize the use of observable inputs and minimize the use of unobservable inputs. The three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value, includes:
| Level 1 — | Observable inputs for identical assets or liabilities such as quoted prices in active markets; | ||||
| Level 2 — | Inputs other than quoted prices in active markets that are either directly or indirectly observable; and | ||||
| Level 3 — | Unobservable inputs in which little or no market data exists, which are therefore developed by the Company using estimates and assumptions that reflect those that a market participant would use. | ||||
On a recurring basis, the Company currently measures and records money market funds (Level 1) and value of Warrants (Level 3) using fair value measurements in the accompanying financial statements. The carrying amounts of the Company's short-term financial instruments, which include cash and cash equivalents, accounts receivable and accounts payable approximate their fair values due to their short maturities.
Significant customers and accounts receivable
Billed accounts receivable are stated at invoice amounts and consist of amounts due from the United States Government ("USG"), commercial and Bioservices customers, as well as amounts due under reimbursement contracts with other government entities and non-government organizations. The Company's branded and generic opioid overdose reversal product, NARCAN® Nasal Spray, is sold commercially over-the-counter at retail pharmacies and digital commerce websites. All branded Naloxone products, including KLOXXADO® Nasal Spray, are sold through physician-directed or standing order prescriptions at retail pharmacies, health departments, local law enforcement agencies, community-based organizations, substance abuse centers and other federal agencies.
91
Concentration Risk
Customers
The Company has long-term contracts with the USG that expire at various times from 2026 through 2036. The Company has derived a significant portion of its revenue from sales of our Government - MCM products under contracts with the USG. The Company's current USG contracts do not necessarily increase the likelihood that it will secure future comparable contracts with the USG. The Company expects that a significant portion of the business will continue to be under government contracts that present a number of risks that are not typically present in the commercial contracting process. USG contracts for ACAM2000® and Anthrax Vaccines and other medical countermeasures products are subject to unilateral termination or modification by the government. The Company may fail to achieve significant sales of its medical countermeasures products, including ACAM2000® and Anthrax Vaccines to customers in addition to the USG, which would harm their growth opportunities. The Company's commercial product sales, largely NARCAN® Nasal Spray, are sold commercially over-the-counter at retail pharmacies and digital commerce websites as well as through physician-directed or standing order prescriptions at retail pharmacies, health departments, local law enforcement agencies, community-based organizations, substance abuse centers and other federal agencies. Refer to Note 14, "Revenue recognition" for more information regarding significant customers.
Although the Company seeks to expand its customer base and to renew its agreements with its customers prior to expiration of a contract, a delay in securing a renewal or a failure to secure a renewal or securing a renewal on less favorable terms may have a material adverse effect on the Company’s financial condition and results of operations.
The Company’s accounts receivable do not represent a significant concentration of credit risk. The USG accounted for approximately 46 %, 39 % and 38 % of total revenues for 2025, 2024 and 2023, respectively. The Company’s accounts receivable as of December 31, 2025 and 2024, consist primarily of amounts due from the USG or other large multi-national highly reputable customers for product sales, Bioservices or from government agencies under government grants.
Financial Institutions
Cash and cash equivalents are maintained with several financial institutions. The Company has deposits held with banks that exceed the amount of insurance provided on such deposits. Generally, these deposits may be redeemed upon demand and are maintained with financial institutions of reputable credit and, therefore, bear minimal credit risk.
Lender Counterparties
There is lender counterparty risk associated with the Company's credit agreement for asset-based revolving loans (the "Revolving Credit Agreement") and any derivative instruments that the Company may utilize from time to time. There is risk that the Company’s Revolving Credit Agreement investors and derivative counterparties will not be available to fund as obligated. If funding under the Revolving Credit Agreement is unavailable, the Company may have to acquire a replacement credit facility from different counterparties at a higher cost or may be unable to find a suitable replacement. The Company seeks to manage risks from its Revolving Credit Agreement and any derivative instruments that the Company may utilize from time to time by contracting with experienced large financial institutions and monitoring the credit quality of its lenders. As of December 31, 2025, the Company does not anticipate nonperformance by any of its counterparties.
92
Inventories, net
Inventories are stated at the lower of cost or net realizable value with cost being determined using a standard cost method, which approximates actual cost. Actual cost consists primarily of material, labor and manufacturing overhead expenses (including fixed production-overhead costs) and includes the services and products of third-party suppliers. For internally manufactured inventory, the Company determines normal capacity for each production facility and allocates fixed production-overhead costs on that basis. The Company records inventory acquired in business combinations utilizing the comparative sales method, which estimates the expected sales price reduced for all costs expected to be incurred to complete/dispose of the inventory with a profit on those costs. The Company analyzes its inventory each reporting period and records a reserve to reduce the cost basis to net realizable value for inventory that has become obsolete, expired, slow-moving or short-dated based on customer requirements. Reserves for excess and obsolete inventory are relieved when the related inventory is disposed of through scrap or sale.
Pre-launch inventory
Within the Company's Commercial Product and MCM Products segments costs relating to raw materials and production of inventory in preparation for product launch prior to regulatory approval are capitalized when the review process has progressed to a point where objective and persuasive evidence exists that regulatory approval is probable, the future economic benefit is expected to be realized, and the Company believes that material uncertainties related to the ultimate regulatory approval have been significantly reduced. Pre-launch inventory is recorded to research and development expense unless these criteria are met. For pre-launch inventory that is capitalized, the Company considers a number of specific facts and circumstances, including the product candidate’s current status in the drug development and regulatory approval process, results from related clinical trials, results from meetings with relevant regulatory agencies prior to the filing of regulatory applications, potential obstacles to the approval process, historical experience, viability of commercialization and market trends. This policy is not applicable to pre-launch inventory purchased to satisfy a performance obligation related to a Bioservices contract as Bioservices pre-launch inventory may be capitalized if it has future economic benefit based on the terms of the contract.
Property, plant and equipment, net
Property, plant and equipment are stated at cost, less accumulated depreciation and impairments. subject to reviews for impairment whenever events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable. The cost of normal, recurring or periodic repairs and maintenance activities related to property, plant and equipment are expensed as incurred. The cost for planned major maintenance activities, including the related acquisition or construction of assets, is capitalized if the repair will result in future economic benefits.
Interest costs incurred during the construction of major capital projects are capitalized until the underlying asset is ready for its intended use, at which point the interest costs are amortized as depreciation expense over the life of the underlying asset.
The Company capitalizes internal-use software when both (a) the software is internally developed, acquired, or modified solely to meet the entity’s internal needs and (b) during the software’s development or modification, no substantive plan either exists or is being developed to market the software externally. Capitalization of qualifying internal-use software costs begins when the preliminary project stage is completed, management with the relevant authority, implicitly or explicitly, authorizes and commits to the funding of the software project, and it is probable that the project will be completed and the software will be used to perform the function intended.
The Company generally depreciates or amortizes the cost of its property, plant and equipment using the straight-line method over the estimated useful lives of the respective assets, which are summarized as follows:
| Land | Not depreciated | ||||
| Buildings | |||||
| Building improvements | |||||
| Furniture and equipment | |||||
| Software | |||||
| Leasehold improvements | Lesser of the asset life or lease term | ||||
Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are removed from the accounts and any resulting gain or loss is credited or charged to operations. Repairs and maintenance costs are expensed as incurred.
93
The Company determines the fair value of the property, plant and equipment acquired in a business combination utilizing either the cost approach or the sales comparison approach. The cost approach is determined by establishing replacement cost of the asset and then subtracting any value that has been lost due to economic obsolescence, functional obsolescence, or physical deterioration. The sales comparison approach determines an asset is equal to the market price of an asset of comparable features such as design, location, size, construction, materials, use, capacity, specification, operational characteristics and other features or descriptions.
Income taxes
Income taxes include federal, state, local and international taxes. Income taxes are accounted for using the asset and liability method. Deferred tax assets/liabilities are recognized for future tax consequences attributable to differences between financial statement carrying amounts of existing assets and liabilities and their respective tax basis and net operating loss and research and development ("R&D") tax credit carryforwards. Deferred tax assets/liabilities are measured using enacted tax rates expected to apply to taxable income in the year in which those temporary differences are expected to be recovered or settled. Valuation allowances are recorded as appropriate to reduce deferred tax assets to the amount considered not more likely than not to be realized.
Deferred income tax effects of transactions reported in different periods for financial reporting and income tax return purposes are recognized under the asset/liability method of accounting for income taxes. This method gives consideration to the future tax consequences of the deferred income tax items and immediately recognizes changes in income tax laws in the year of enactment.
The Company's ability to realize deferred tax assets depends upon future taxable income as well as the limitations discussed below. For financial reporting purposes, a deferred tax asset must be reduced by a valuation allowance if it is not more likely than not that some portion or all of the deferred tax assets will be realized. In general, if the Company determines that it is more likely than not to realize more than the recorded amounts of net deferred tax assets in the future, the Company will reverse all or a portion of the valuation allowance established against its deferred tax assets, resulting in a decrease to income taxes in the period in which the determination is made. Likewise, if the Company determines that it is not more likely than not to realize all or part of the net deferred tax asset in the future, the Company will establish a valuation allowance against deferred tax assets, with an offsetting increase to income taxes, in the period in which the determination is made.
Under sections 382 and 383 of the Internal Revenue Code, if an ownership change occurs with respect to a "loss corporation", as defined therein, there are annual limitations on the amount of net operating losses and credits that are available. Historically, the Company has recognized the portion of net operating losses and R&D tax credits acquired that will not be limited and are more likely than not to be realized.
Because tax laws are complex and subject to different interpretations, significant judgment is required. As a result, the Company makes certain estimates and assumptions, in (1) calculating the Company's income tax expense, deferred tax assets/liabilities, (2) determining any valuation allowance recorded against deferred tax assets and (3) evaluating the amount of unrecognized tax benefits, as well as the interest and penalties related to such uncertain tax positions. The Company's estimates and assumptions may differ from tax benefits ultimately realized.
Asset Impairment Analysis
Goodwill and Indefinite-lived Intangible Assets
Goodwill represents the difference between the purchase price and the fair value of the identifiable tangible and intangible net assets when accounted for using the purchase method of accounting. Goodwill is not amortized but is reviewed for impairment. Goodwill is allocated to the Company's reporting units, which are components of our business for which discrete cash flow information is available one level below its operating segment. The Company evaluates goodwill and other indefinite-lived intangible assets for impairment annually as of October 1 and at interim times if an event or other circumstance indicates that we may not recover the carrying value of the asset. If the Company believes that as a result of its qualitative assessment it is more likely than not that the fair value of a reporting unit or other indefinite-lived intangible asset is greater than its carrying amount, the quantitative impairment test is not required. If however it is determined that it is not more likely than not that the fair value of a reporting unit or other indefinite-lived intangible asset is greater than its carrying amount, a quantitative test is required.
The quantitative goodwill impairment test is performed using a one-step process. The process is to compare the fair value of a reporting unit with its carrying amount. If the fair value of a reporting unit exceeds its carrying amount, goodwill of the reporting unit is not impaired. If the carrying amount of a reporting unit exceeds its fair value, goodwill of the reporting unit is impaired and an impairment loss is recognized in an amount equal to that excess up to the total amount of goodwill included in the reporting unit.
94
When the Company has material indefinite lived intangible assets associated with in-process research and development ("IPR&D"), a qualitative assessment is performed. If the qualitative assessment indicates that it is not more likely than not that the fair value of the indefinite lived intangible asset exceeds its carrying amount, the Company compares the estimated fair value of the intangible with its carrying value. If the carrying value of the intangible asset exceeds its fair value, an impairment loss is recognized in an amount equal to that excess. Determining fair value requires the exercise of judgment about appropriate discount rates, perpetual growth rates and the amount and timing of expected future cash flows (see Note 7, "Intangible assets and goodwill"). Upon successful completion of each project, the asset is classified as a definite-lived intangible and the Company will decide as to the then-useful life of the intangible asset, generally determined by the period in which the substantial majority of the cash flows are expected to be generated and begin amortization.
Long-lived Assets
Long-lived assets such as intangible assets and property, plant and equipment are not required to be tested for impairment annually. Instead, they are tested for impairment whenever circumstances indicate that the carrying amount of the asset may not be recoverable, such as when the disposal of such assets is likely or there is an adverse change in the market involving the business employing the related assets. If an impairment analysis is required, the impairment test employed is based on whether the Company’s intent is to hold the asset for continued use or to hold the asset for sale. If the intent is to hold the asset for continued use, the impairment test first requires a comparison of undiscounted future cash flows to the carrying value of the asset. If the carrying value of the asset exceeds the undiscounted cash flows, the asset would not be deemed to be recoverable. Impairment would then be measured as the excess of the asset’s carrying value over its fair value. Fair value is typically determined by discounting the future cash flows associated with that asset. If the intent is to hold the asset for sale and certain other criteria are met, the impairment test involves comparing the asset’s carrying value to its fair value less costs to sell. To the extent the carrying value is greater than the asset’s fair value less costs to sell, an impairment loss is recognized in an amount equal to the difference. Significant judgments used for long-lived asset impairment assessments include identifying the appropriate asset groupings and primary assets within those groupings, determining whether events or circumstances indicate that the carrying amount of the asset may not be recoverable, determining the future cash flows for the assets involved and assumptions applied in determining fair value, which include, reasonable discount rates, growth rates, market risk premiums and other assumptions about the economic environment.
Contingent Consideration
The Company's acquisitions accounted for as asset acquisitions may include contingent consideration payments to be made for sales-based royalties, sales-based milestones and development and regulatory milestones. The Company assesses whether such contingent consideration meets the definition of a derivative. Contingent consideration payments in an asset acquisition not required to be accounted for as derivatives are recognized when the contingency is resolved, and the consideration is paid or becomes payable. Contingent consideration payments required to be accounted for as derivatives are recorded at fair value on the date of the acquisition and are subsequently remeasured to fair value at each reporting date.
Leases
The Company has operating leases for corporate offices, R&D facilities and manufacturing facilities. The Company determines if an arrangement is a lease at inception. Operating leases with future minimum lease payments in excess of 12 months are included in right-of-use ("ROU") assets and liabilities. The Company has elected to record expense as incurred for leases with minimum lease payments of 12 months or less.
ROU assets represent the Company's right to use an underlying asset for the lease term and lease liabilities represent the Company's obligation to make lease payments arising from the lease. Operating lease ROU assets and liabilities are recognized at commencement date based on the present value of lease payments over the lease term. As most of the Company's leases do not provide an implicit rate, the Company uses an incremental borrowing rate based on the information available at commencement date in determining the present value of lease payments. The Company uses an implicit rate when readily determinable. At the beginning of a lease, the operating lease ROU asset also includes any concentrated lease payments expected to be paid and excludes lease incentives. The Company's lease ROU asset may include options to extend or terminate the lease when it is reasonably certain that the Company will exercise those options.
Lease expense for lease payments is recognized on a straight-line basis over the lease term. The Company has lease agreements with lease and non-lease components, which are accounted for separately.
95
Revenue recognition
The Company recognizes revenue when the Company's customers obtain control of promised goods or services, in an amount that reflects the consideration which the Company expects to receive in exchange for those goods or services by analyzing the following five steps: (1) identify the contract with a customer(s); (2) identify the performance obligations in the contract; (3) determine the transaction price; (4) allocate the transaction price to the performance obligations in the contract; and (5) recognize revenue when (or as) the entity satisfies a performance obligation.
Multiple performance obligations
At contract inception, the Company assesses the products or services promised in a contract and identifies a performance obligation for each promise to transfer to the customer a product or service that is distinct, including evaluating whether the contract includes a customer option for additional goods or services which could represent a material right. A performance obligation is a promise in a contract to transfer a distinct product or service to a customer and is the unit of account under ASC 606. Contracts sometimes include more than one product, a lease, or options for customers to purchase additional products or services in the future for free or at a discount, which gives rise to separate performance obligations. For contracts with multiple performance obligations, the Company allocates the contract price to each performance obligation on a relative standalone selling price basis using the Company’s best estimate of the standalone selling price of each distinct product or service in the contract. The primary method used to estimate standalone selling price is the price observed in standalone sales to customers, however when prices in standalone sales are not available the Company may use third-party pricing for similar products or services or estimate the standalone selling price. Allocation of the transaction price is determined at the contracts’ inception.
Transaction price and variable consideration
Once the performance obligations in the contract have been identified, the Company estimates the transaction price of the contract. The estimate includes amounts that are fixed as well as those that can vary based on expected outcomes of the activities or contractual terms. The Company's variable consideration includes net profit received from sales of the Company's Naloxone products, certain MCM products sold on a net basis, cost-plus-fee contract terms and consideration transferred under its development contracts as consideration received can vary based on developmental progression of the product candidate. When a contract's transaction price includes variable consideration, the Company evaluates the variable consideration to determine whether the estimate needs to be constrained; therefore, the Company includes the variable consideration in the transaction price only to the extent that it is probable that a significant reversal of the amount of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. Variable consideration estimates are updated at each reporting date. There were no significant constraints or material changes to the Company's variable consideration estimates as of or during the year ended December 31, 2025.
Product sales
For our product sales, we recognize revenue at a point in time when the Company’s performance obligations have been satisfied and control of the products transfer to the customer. To indicate the transfer of control the Company will have a present right to payment, legal title must have passed to the customer, and the customer must have the significant risks and rewards of ownership. This point in time depends on several factors, including delivery, transfer of legal title, transition of risk and rewards of the product to the customer and the Company's right to payment.
The Company's contracts for the sale of the Company's Government - MCM products include certain acceptance criteria before title passes to the customer. The primary customer for the Company's Government - MCM products and the primary source of funding for the development of its MCM product candidate portfolio is the USG. The USG contracts for the sale of the Company's Government - MCM products are normally multi-year contracts with annual options. MCM product revenue is recognized to the extent that it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur.
96
The Company's Nasal Naloxone Products are sold commercially over-the-counter at retail pharmacies and digital commerce websites as well as through physician-directed or standing order prescriptions at retail pharmacies, health departments, local law enforcement agencies, community-based organizations, substance abuse centers and other federal agencies.
The Company's OTC NARCAN® customer contracts are fixed price contracts. The Company invoices and records revenue when the pharmacies and wholesalers receive product from the third-party logistics warehouse used by the Company, which is the point at which control is transferred to the customer. Revenues for OTC NARCAN® are recorded at the net sales price (transaction price), which includes estimates of variable consideration for which reserves are established. Estimates of variable consideration include allowance for returns, specialty distributor fees, wholesaler fees and prompt payment discounts. OTC NARCAN® may also be sold on consignment through third-party online retailers where revenues are recognized at the point in time when sold to the end customer. The Company pays these third-party online retailers selling commissions and fulfillment fees which are recorded as SG&A expenses and Cost of Commercial Product sales, respectively, in the Consolidated Statement of Operations. Revenues from OTC NARCAN® are recognized to the extent that it is probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with such variable consideration is subsequently resolved. The Company considers several factors in the estimation process for the allowance for returns of OTC NARCAN®, including inventory levels within the distribution channel, product shelf life and historical return activity, including activity for product sold for which the return period has passed, as well as other relevant factors. Because returned product cannot be resold, there is no corresponding asset for product returns.
Bioservices
The Company performs it's Bioservices offerings for third parties. Under these contracts, activities can include drug substance and drug product manufacturing services for injectable and other sterile products, and development services such as pharmaceutical product process development, process design, technology transfer, manufacturing validations, laboratory analytical development support, aseptic filling, lyophilization, final packaging, stability studies, and suite-reservations. These contracts vary in duration, activities, and number of performance obligations. Performance obligations identified under these arrangements may include drug substance and/or drug product manufacturing, technology transfer activities, and suite-reservations.
Drug substance, drug product manufacturing, development services and technology transfer performance obligations are recognized as revenue over-time because the Company’s performance does not create an asset with an alternative use and the Company has an enforceable right to payment for performance completed as work is performed. In drug product arrangements, the customer typically owns and supplies the active pharmaceutical ingredient (API), that is used in the manufacturing process; in drug substance arrangements, the customer provides certain seed material that is used in the manufacturing process. The transaction price generally contains both a fixed and variable component. The fixed component is stated in the agreement as a fixed price per unit with no contractual provision for a refund or price concession and the variable component generally results from pass-through costs that are billed at cost-plus over the life of the contract. The Company uses an input method to measure progress toward the satisfaction of the related performance obligations based on costs incurred as a percentage of total costs to complete which the Company believes best depicts the transfer of control of goods or services promised to its customers.
Suite reservations are classified as leases when the customer directs the use of the identified suite and obtains substantially all the economic benefits from the manufacturing capacity. If a customer reserves more than one suite, the allocation of contract value is based on relative selling price which varies due to size, location, capacity, production capability for drug product or drug substance, and the time of planned use. The associated revenue is recognized on a straight-line basis over the period of performance. For arrangements that contain both lease and non-lease components, consideration in the contract is allocated on a relative standalone selling price basis.
The Company’s Bioservices customer contracts generally include provisions entitling the Company to a termination penalty when the contract is terminated prior to the contract’s nominal end date. The termination penalties in the customer contracts vary but are generally considered substantive for accounting purposes and create enforceable rights and obligations throughout the stated duration of the contract. The Company accounts for a contract cancellation as a contract modification. The determination of the contract termination penalty is based on the terms stated in the related customer agreement. As of the modification date, the Company updates its estimate of the transaction price, subject to constraints, and recognizes the amount over the remaining performance period or measure of progress under the arrangement.
For contracts that contain lease components, the Company assesses the collectability of the lease payments. If the collectability of the lease payments is probable, the Company recognizes lease income over the term of the lease on a straight-line basis. If collectability is not deemed probable at any time during the term of the lease, the Company’s lease income is limited to the lesser of (i) the lease payments that have been collected from the lessee, or the straight-line recognition of the contract value. If the collectability assessment changes to probable after the Company has determined collectability is not deemed probable, any difference between the lease income that would have been recognized if collectability had always been assessed as probable and the lease income recognized to date is recognized as a current-period adjustment to lease income. Changes to the collectability of operating leases are recorded as adjustments to lease income in the Consolidated Statements of Operations in the period that they occur.
97
Contracts and grants
Research and development
The Company expenses R&D costs as incurred. The Company's R&D expenses consist primarily of:
▪personnel-related expenses;
▪fees to professional service providers for, among other things, analytical testing, independent monitoring or other administration of the Company's clinical trials and obtaining and evaluating data from the Company's clinical trials and non-clinical studies;
▪costs associated with technology transfer and scale up activities throughout the development stage, including internally and through third party contract manufacturers;
▪costs of Bioservices for clinical trial material; and
▪costs of materials intended for use and used in clinical trials and R&D.
Comprehensive income (loss)
Comprehensive income (loss) is comprised of net income (loss) and other changes in equity that are excluded from net income (loss). The Company includes translation gains and losses incurred when converting its subsidiaries' financial statements from their functional currency to the U.S. dollar in accumulated other comprehensive income (loss) as well as gains and losses on its pension benefit obligation and derivative instruments.
Translation and remeasurement of foreign currencies
For our non-U.S. subsidiaries that transact in a functional currency other than the U.S. dollar, assets and liabilities are translated at current rates of exchange at the balance sheet date. Income and expense items are translated at the average foreign currency exchange rates for the period. Adjustments resulting from the translation of the financial statements of our foreign operations into U.S. dollars are excluded from the determination of net income (loss) and are recorded in accumulated other comprehensive income (loss), a separate component of equity. For subsidiaries where the functional currency of the assets and liabilities differ from the local currency, non-monetary assets and liabilities are remeasured at the rate of exchange in effect on the date assets were acquired while monetary assets and liabilities are remeasured at current rates of exchange as of the balance sheet date. Income and expense items are remeasured at the average foreign currency rates for the period. Remeasurement adjustments of these subsidiaries are included in "Other income (expense), net" in our Consolidated Statements of Operations.
Earnings (loss) per common share
Treasury stock
When stock is acquired for purposes other than formal or constructive retirement, the purchase price of the acquired stock is recorded in a separate treasury stock account, which is separately reported as a reduction of equity.
When stock is retired or purchased for formal or constructive retirement, the purchase price is initially recorded as a reduction to the par value of the shares repurchased, with any excess purchase price over par value recorded as a reduction to additional paid-in capital related to the series of shares repurchased and any remainder excess purchase price recorded as a reduction to retained earnings. If the purchase price exceeds the amounts allocated to par value and additional paid-in capital related to the series of shares repurchased and retained earnings, the remainder is allocated to additional paid-in capital related to other series of shares.
98
To determine the cost of treasury stock that is either sold or reissued, the Company uses the last in, first out method. If the proceeds from the re-issuance of treasury stock are greater than the cost, the excess is recorded as additional paid-in capital. If the proceeds from re-issuance of treasury stock are less than the cost, the excess cost first reduces any additional paid-in capital arising from previous treasury stock transactions for that class of stock, and any additional excess is recorded as a reduction of retained earnings.
Advertising Costs
Accounting for share-based compensation
The Company maintains the Fourth Amended and Restated Emergent BioSolutions Inc. 2006 Stock Incentive Plan (the “Emergent Plan”) which provides for grants of equity awards, including stock options, performance stock options, restricted stock units and performance stock units. The Emergent BioSolutions Inducement Plan was maintained by the Company until its termination on December 30, 2025. Following its termination, no new awards will be granted under the Inducement Plan; however, outstanding awards remain subject to its original terms. The terms and conditions of the Inducement Plan are substantially similar to the Company's stockholder-approved Emergent Plan as discussed below. For all of our share-based awards, the Company recognizes forfeitures and compensation costs when they occur.
The terms and conditions of equity awards (such as price, vesting schedule, term and number of shares) under the Emergent Plan is determined by the compensation committee of the Company's board of directors, which administers the Emergent Plan. Each equity award granted under the Emergent Plan vests as specified in the relevant agreement with the award recipient and no option can be exercised after seven years from the date of grant. The Company records the estimated fair value of awards in expense on a straight-line basis over the requisite service period, which is generally the vesting period. Where awards are made with non-substantive vesting periods (for instance, where a portion of the award vests upon retirement eligibility), the Company estimates and recognizes expense based on the period from the grant date to the date the employee becomes retirement eligible.
The Company determines the fair value of restricted stock units using the closing market price of the Company's common stock on the day prior to the date of grant. The Company's performance stock units settle in the Company's stock. The fair value is determined on the date of the grant using the number of shares expected to be earned and the ending market value of the stock on the day prior to the grant date. The number of shares expected to vest is adjusted each reporting period by assessing the probability that the performance criteria will be met and the associated targeted payout level that is forecasted will be achieved.
The Company utilizes the Black-Scholes valuation model for estimating the fair value of stock options granted. Set forth below is a discussion of the Company's methodology for developing each of the assumptions used:
▪Expected dividend yield — the Company does not pay regular dividends on its common stock and does not anticipate paying any dividends in the foreseeable future.
▪Expected volatility — a measure of the amount by which a financial variable, such as share price, has fluctuated (historical volatility) or is expected to fluctuate (implied volatility) during a period. The Company analyzed its own historical volatility to estimate expected volatility over the same period as the expected average life of the options.
▪Risk-free interest rate — the range of U.S. Treasury rates with a term that most closely resembles the expected life of the option as of the date on which the option is granted.
▪Expected average life of options — the period of time that options granted are expected to remain outstanding, based primarily on the Company's expectation of option exercise behavior subsequent to vesting of options.
The Company uses a Monte Carlo valuation model for estimating performance stock options and liability classified awards granted that vest based on market conditions. The Monte Carlo model incorporates more complex variables than closed-form models such as the Black-Scholes valuation model used for stock option grants. The Monte Carlo valuation model simulates a distribution of stock prices to yield an expected distribution of stock prices over the performance period. The stock-paths are simulated using volatilities calculated with historical information using data from a look-back period that is equal to the term of the award. The model assumes a risk-free interest rate with a term equal to the expected term of the award. The simulations are repeated multiple times and the mean of the discounted values is calculated as the grant date fair value for the award. The final payout of the award as calculated by the model is then discounted back to the grant date using the risk-free interest rate.
99
New Accounting Standards
Recently Adopted Accounting Standards
In December 2023, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update ("ASU") No. 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures. The amendment enhances income tax disclosure requirements by requiring public entities to provide additional disaggregation in the income tax rate reconciliation and to disclose income taxes paid by jurisdiction. The guidance is effective for annual reporting periods in fiscal years beginning after December 15, 2024. The Company elected to adopt ASU 2023-09 in accordance with its effective date and will apply the guidance prospectively. Beginning with the current reporting period, the Company has enhanced its income tax disclosures included in Note 16, "Income taxes", to comply with the new requirements.
Accounting Standards Not Yet Adopted
In November 2024, the FASB issued ASU 2024-03, Income Statement - Reporting Comprehensive Income - Expense Disaggregation Disclosures (Subtopic 220-40) Disaggregation of Income Statement Expenses, which requires a public business entity to disclose additional information about specific expense categories in the notes to financial statements on an annual and interim basis. The amendments are effective for annual periods beginning after December 15, 2026, and interim periods beginning after December 15, 2027, with early adoption permitted. A public entity should apply the amendments either prospectively to financial statements issued for reporting periods after the effective date of this ASU or retrospectively to any or all prior periods presented in the financial statements. The Company is in the process of evaluating the impact of this new guidance on its consolidated financial statements.
In December 2025, the FASB issued ASU 2025-10, Government Assistance (Topic 832): Accounting for Government Grants Received by Business Entities. This guidance establishes authoritative requirements for recognition, measurement, presentation, and disclosure of government grants received by business entities. The ASU aligns U.S. GAAP with certain aspects of IAS 20 and requires entities to disclose the nature and terms of government grants, significant conditions, accounting policies applied, amounts recognized in the financial statements, and any repayment obligations. ASU 2025-10 is effective for public business entities for annual periods beginning after December 15, 2028, including interim periods within those fiscal years, with early adoption permitted. The Company is currently evaluating the impact of this guidance on its consolidated financial statements and related disclosures. Adoption is expected to require enhanced disclosures regarding government grants received and may affect the timing of income recognition for certain arrangements. The Company does not expect the adoption of ASU 2025-10 to have a material impact on its financial position or results of operations.
3. Assets and liabilities held-for-sale
In March 2025, the Company classified the assets and related liabilities associated with warehouse space in Maryland as held-for-sale. At that time, the Company recognized a non-cash loss on assets held-for-sale of $12.2 million which adjusted the disposal group to their fair values less costs to sell. During the fourth quarter of 2025, the Company concluded that its Maryland warehouse no longer met the criteria to be classified as held-for-sale due to changes in management's plan regarding the use of the warehouse space and determined it was no longer probable that it would be sold within 12 months. The net assets were reclassified to held-and-used at the lower of (i) their carrying amount, adjusted for depreciation that would have been recognized had they remained classified as held-and-used, or (ii) their fair value as of the date the decision not to sell was made. This reclassification as of December 31, 2025 did not result in any incremental adjustments to the recorded value of the assets and liabilities and is reported within “Impairment of long-lived assets” on the Consolidated Statements of Operations for the year ended December 31, 2025.
4. Divestitures
Sale of Travel Health Business
In May 2023, pursuant to the Purchase and Sale Agreement (the “Purchase and Sale Agreement”), by and between the Company, through its wholly owned subsidiaries Emergent International Inc. and Emergent Travel Health Inc., and Bavarian Nordic (“Bavarian Nordic”), the Company completed the previously announced sale of the Company’s travel health business, including rights to Vivotif®, the licensed typhoid vaccine; Vaxchora®, the licensed cholera vaccine; the development-stage chikungunya vaccine candidate CHIKV VLP; the Company’s manufacturing site in Bern, Switzerland; and certain of its development facilities in San Diego, California.
At the closing, Bavarian Nordic paid a cash purchase price of $270.2 million, exclusive of customary closing adjustments for cash, indebtedness, working capital and transaction expenses of the business at closing. Bavarian Nordic was required to pay milestone payments totaling $80.0 million, which the Company recorded within “Other, net” in the Consolidated Statement of Operations, upon satisfaction of the following milestones:
100
•In July 2024, Bavarian Nordic announced that the European Medicines Agency had validated the marketing authorization application for CHIKV VLP, which triggered a development milestone payment receivable in the amount of $10.0 million. The Company received this milestone payment in the fourth quarter of 2024.
•In August 2024, Bavarian Nordic announced that the FDA has accepted and granted Priority Review for the Biologics License Application for CHIKV VLP, which triggered a milestone payment receivable in the amount of $20.0 million. The Company received this milestone payment in the fourth quarter of 2024.
•In February 2025, Bavarian Nordic announced that the FDA approved CHIKV VLP under the Priority Review, which triggered a milestone payment in the amount of $30.0 million which was recorded in “Other, net” during the year ended December 31, 2025. The Company received this milestone payment during the first quarter of 2025.
•In February 2025, Bavarian Nordic announced that the European Commission approved CHIKV VLP, which triggered a milestone payment receivable in the amount of $20.0 million which was recorded in “Other, net” during the year ended December 31, 2025. The Company received this milestone payment during the second quarter of 2025.
Pursuant to the Purchase and Sale Agreement, the Company may receive up to $30.0 million of earn-out payments from Bavarian Nordic based on aggregate net sales of Vaxchora® and Vivotif® in calendar year 2026.
Sale of RSDL®
In July 2024, the Company, through its wholly owned subsidiary Emergent BioSolutions Canada Inc., entered into the RSDL® Agreement with SERB pursuant to which, among other things, the Company sold its worldwide rights to RSDL® to SERB. The RSDL® Transaction also included the sale to SERB of all the outstanding capital stock of Emergent Protective Products USA Inc. (“EPPU”), a wholly owned subsidiary of the Company, which leases a manufacturing facility in Hattiesburg, Mississippi, as well as certain assets related to RSDL®, including intellectual property rights, contract rights, inventory and marketing authorizations. In addition, the employees of EPPU joined SERB in connection with the RSDL® Transaction.
At the closing, SERB paid a cash purchase price of $75.0 million, exclusive of customary closing adjustments related to inventory. In addition, SERB will owe the Company a $5.0 million payment upon achievement of a milestone relating to sourcing of a certain component of RSDL® decontamination lotion. In connection with the RSDL® Transaction, the Company recognized a pre-tax gain of $60.8 million, net of transaction costs of $4.1 million, recorded within “Gain on sale of business” on the Consolidated Statements of Operations during the year ended December 31, 2024.
The Company and SERB entered into a transition services agreement (the “SERB TSA”) to ensure the orderly transition of RSDL® decontamination lotion and the related assets to SERB, and a supply agreement (the “SERB Supply Agreement”) pursuant to which SERB has a suite reservation at the Company’s Winnipeg facility where the Company will perform Bioservices activities to manufacture and supply bulk lotion to SERB. The Company and SERB also entered into a reverse supply agreement (together with the SERB TSA and the SERB Supply Agreement, the “SERB Agreements”) pursuant to which SERB supplies to the Company finished RSDL® for the purposes of the Company’s performance of certain transitional distribution services under customer contracts that have not yet transferred to SERB. Under the SERB Agreements, the Company will retain a portion of net sales received upon delivery of RSDL® to the delayed transfer customers. Income from performing services under the SERB TSA is recorded within “Product and services sales, net” on the Consolidated Statements of Operations and was $1.9 million for the year ended December 31, 2025.
Sale of Baltimore-Camden Facility
In August 2024, pursuant to the Asset Purchase Agreement, the Company completed the sale of its Drug Product facility in Baltimore-Camden to an affiliate of Bora. The Baltimore-Camden facility, which was part of the Company’s Services operating segment, had clinical and commercial non-viral aseptic fill/finish services on four fill lines, including lyophilization, formulation development, and support services. At closing, Bora paid a cash purchase price of approximately $35.0 million, which included customary closing adjustments to date for working capital and transaction expenses of the business at closing. As a result of the divestiture, the Company recognized a pre-tax loss of $36.5 million, net of transaction costs of $3.8 million, during the year ended December 31, 2024 recorded within “Gain on sale of business” on the Consolidated Statements of Operations.
In connection with the divestiture, the Company entered into a Transition Services Agreement (the “Bora TSA”) with Bora to help support its ongoing operations. Under the Bora TSA, the Company is providing certain transition services to Bora, including information technology, finance and enterprise resource planning, human resources, employee benefits and other limited services. Income from performing services under the Bora TSA is recorded within “Other, net” on the Consolidated Statements of Operations and was $0.4 million for the year ended December 31, 2025.
101
Sale of Baltimore-Bayview Facility
In March 2025, the Company completed the sale of its Baltimore-Bayview drug substance manufacturing facility to Syngene International (“Syngene”). At closing, Syngene paid a cash purchase price of $36.5 million. Pursuant to the sale, Syngene acquired the assets and equipment associated with the Baltimore-Bayview facility.
As a result of the divestiture, the Company recognized a pre-tax gain of $7.9 million, net of transaction costs of $1.2 million, during the year ended December 31, 2025, recorded within “Other, net” on the Consolidated Statements of Operations. The Company determined that the disposal of the Baltimore-Bayview facility does not qualify for reporting as a discontinued operation since it does not represent a strategic shift that has or will have a major effect on the Company’s operations and financial results.
5. Impairment and restructuring charges
2025 Impairment of long-lived assets
The Company recognized a $12.2 million non-cash loss on assets held-for-sale classification during 2025 related to the write‑down of certain Maryland warehouse assets to fair value less cost to sell. During the fourth quarter of 2025, the Company concluded that its Maryland warehouse no longer met the criteria to be classified as held-for-sale and reclassified. This reclassification affected only the presentation of the original charge and did not result in any incremental impairment or impact on the Company’s results of operations. For further information regarding the classification change, refer to Note 3, "Assets and liabilities held-for-sale".
2024 Impairment of long-lived assets
During the preparation of the Company’s financial statements for the three months ended June 30, 2024, due to the decision to close the Company’s Baltimore-Bayview Drug Substance manufacturing facility and the Rockville, Maryland Drug Product facility, the Company determined there were sufficient indicators of impairment for the Bayview and Rockville asset groups within the Bioservices reporting unit. As a result, the Company performed recoverability tests on those asset groups and concluded that the Bayview and Rockville asset groups were not recoverable as the undiscounted expected cash flows did not exceed their carrying values.
Asset groups are written down only to the extent that their carrying value is higher than their respective fair value. The Company, with the assistance of a third-party valuation firm, applied valuation methods to estimate the fair values for each of the assets within the different asset classes. An orderly liquidation value was applied to estimate the fair value of the personal property assets and market and cost based approaches were applied to estimate the fair value of the real property assets, each representing Level 3 non-recurring fair value measurements. Based on these analyses, the Company allocated and recognized a non-cash impairment charge of $27.2 million during the second quarter of 2024.
2023 Impairment of long-lived assets
During the preparation of the Company’s financial statements for the three months ended June 30, 2023, due to deterioration in performance and resulting downward revisions to the Company’s internal Bioservices forecast made during the second quarter, including future expected cash flows, the Company determined there were sufficient indicators of impairment on the Camden, Bayview and Rockville asset groups within the Bioservices reporting unit to require an impairment analysis. As a result, the Company performed recoverability tests on certain asset groups within the Bioservices reporting unit and concluded that the impacted asset groups were not recoverable as the undiscounted expected cash flows did not exceed their carrying values.
Asset groups are written down only to the extent that their carrying value is higher than their respective fair value. The Company, with the assistance of a third-party valuation firm, applied valuation methods to estimate the fair values for each of the assets within the different asset classes. An orderly liquidation value was applied to estimate the fair value of the personal property assets and market and cost based approaches were applied to estimate the fair value of the real property assets, each representing Level 3 non-recurring fair value measurements. Based on these analyses, the Company allocated and recognized a non-cash impairment charge of $306.7 million during the second quarter of 2023.
102
The table below presents the total impairment charge by asset class for the years ended December 31, 2025, 2024, and 2023:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Buildings, building improvements and leasehold improvements | $ | $ | $ | ||||||||||||||
| Furniture and equipment | |||||||||||||||||
| Software | |||||||||||||||||
| Construction-in-progress | |||||||||||||||||
| Total impairment of long-lived assets | $ | $ | $ | ||||||||||||||
Restructuring Charges
January 2023 Organizational Restructuring Plan
In January 2023, the Company initiated an organizational restructuring plan (the “January 2023 Plan”) intended to reduce operating costs, improve operating margins, and continue advancing the Company’s ongoing commitment to profitable growth. As part of the January 2023 Plan, the Company reduced its workforce by approximately 125 employees. The charges related to the January 2023 Plan consisted primarily of charges related to employee transition, severance payments and employee benefits. The cumulative amount of restructuring charge related to the January 2023 Plan since inception is $9.3 million. All activities related to the January 2023 Plan were substantially completed during the first quarter of 2023. Restructuring costs (benefits) are recognized as an operating expense within the Consolidated Statement of Operations and are classified based on the Company's classification policy for each category of operating expense.
August 2023 Organizational Restructuring Plan
In August 2023, the Company initiated the August 2023 Plan, which was intended to strengthen its core business and financial position by reducing investment in and de-emphasizing focus on its CDMO services business for future growth. As part of the August 2023 Plan, the Company reduced its workforce by approximately 400 employees. The charges related to the August 2023 Plan consisted primarily of employee transition, severance payment and employee benefit charges. The cumulative amount of restructuring charge related to the August 2023 Plan since inception is $19.4 million. All activities related to the August 2023 Plan were substantially completed during the third quarter of 2023. Restructuring costs (benefits) are recognized as an operating expense within the Consolidated Statement of Operations and are classified based on the Company’s classification policy for each category of operating expense.
May 2024 Organizational Restructuring Plan
In May 2024, the Company initiated the May 2024 Plan. These strategic actions led to a reduction of the Company’s workforce by approximately 300 employees across all areas of the Company and the elimination of approximately 85 positions that were vacant, as well as the closure of the Company’s Baltimore-Bayview Drug Substance manufacturing facility and Rockville, Maryland Drug Product facility. Decisions regarding the elimination of positions and the closure of manufacturing facilities were subject to local law and consultation requirements in certain countries, as well as the Company’s business needs. The cumulative amount of restructuring charge related to the May 2024 Plan since inception is $18.5 million. All activities related to the May 2024 Plan were substantially completed during the third quarter of 2024. Restructuring costs (benefits) are recognized as an operating expense within the Consolidated Statement of Operations and are classified based on the Company's classification policy for each category of operating expense.
August 2024 Organizational Restructuring Plan
In August 2024, the Company initiated an organizational restructuring plan ("the August 2024 Plan") at the Company’s Lansing facility, which reduced the Company’s workforce by approximately 70 employees, as well as eliminated several open positions. The Company also implemented non-labor optimization efforts, such as reducing the Company’s external and vendor spend. The cumulative amount of restructuring charges related to the August 2024 Plan since inception is $2.5 million. All activities related to the August 2024 Plan were substantially completed during the fourth quarter of 2024. Restructuring costs (benefits) are recognized as an operating expense within the Consolidated Statement of Operations and are classified based on the Company’s classification policy for each category of operating expense.
103
The following table presents the total restructuring costs (benefits) related to the January 2023 Plan, August 2023 Plan, May 2024 Plan and August 2024 Plan by reportable segment as well as amounts included within non-reportable segments, unallocated corporate selling, general and administrative (“SG&A”) expense and research and development (“R&D”) expense:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
Segment restructuring costs (benefits): | |||||||||||||||||
| MCM Products | $ | ( | $ | $ | |||||||||||||
| All other segments | |||||||||||||||||
Total restructuring costs (benefits) included in Cost of product and services sales, net | $ | ( | $ | $ | |||||||||||||
Corporate restructuring costs (benefits): | |||||||||||||||||
| SG&A | $ | ( | $ | $ | |||||||||||||
| R&D | ( | ||||||||||||||||
| $ | ( | $ | $ | ||||||||||||||
The following table presents the total restructuring costs (benefits), by function, for the years ended December 31, 2025, 2024, and 2023:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Employee transition | $ | $ | $ | ||||||||||||||
| Severance payments | ( | ||||||||||||||||
| Employee benefits | ( | ||||||||||||||||
Total restructuring costs (benefits) | $ | ( | $ | $ | |||||||||||||
All financial impacts of the January 2023 Plan were reflected in the Company’s consolidated financial statements by the second quarter of 2024. As a result, there was no activity related to the January 2023 Plan for the year ended December 31, 2025. The following table provides the components of and changes in the Company’s restructuring accrual for the January 2023 Plan during the year ended December 31, 2024:
| Employee Transition | Severance Payments | Employee Benefits | Total | ||||||||||||||||||||
| Balance at December 31, 2023 | $ | $ | $ | $ | |||||||||||||||||||
| Accruals | |||||||||||||||||||||||
| Cash payments | ( | ( | |||||||||||||||||||||
| Balance at December 31, 2024 | $ | $ | $ | $ | |||||||||||||||||||
104
All financial impacts of the August 2023 Plan were reflected in the Company’s consolidated financial statements by the fourth quarter of 2024. As a result, there was no activity related to the August 2023 Plan for year ended December 31, 2025. The following table provides the components of and changes in the Company’s restructuring accrual for the August 2023 Plan during the year ended December 31, 2024:
| Employee Transition | Severance Payments | Employee Benefits | Total | ||||||||||||||||||||
| Balance at December 31, 2023 | $ | $ | $ | $ | |||||||||||||||||||
| Accruals | ( | ( | |||||||||||||||||||||
| Cash payments | ( | ( | ( | ||||||||||||||||||||
| Balance at December 31, 2024 | $ | $ | $ | $ | |||||||||||||||||||
The following table provides the components of and changes in the Company's restructuring accrual for the May 2024 Plan during the years ended December 31, 2025 and 2024:
| Employee Transition | Severance Payments | Employee Benefits | Total | ||||||||||||||||||||
| Balance at December 31, 2023 | $ | $ | $ | $ | |||||||||||||||||||
| Accruals | |||||||||||||||||||||||
| Cash payments | ( | ( | ( | ( | |||||||||||||||||||
| Balance at December 31, 2024 | $ | $ | $ | $ | |||||||||||||||||||
| Accruals | ( | ( | ( | ||||||||||||||||||||
| Cash payments | ( | ( | ( | ||||||||||||||||||||
| Balance at December 31, 2025 | $ | $ | $ | $ | |||||||||||||||||||
The following table provides the components of and changes in the Company's restructuring accrual for the August 2024 Plan during the year ended December 31, 2025:
| Employee Transition | Severance Payments | Employee Benefits | Total | ||||||||||||||||||||
| Balance at December 31, 2023 | $ | $ | $ | $ | |||||||||||||||||||
| Accruals | |||||||||||||||||||||||
| Cash payments | ( | ( | ( | ( | |||||||||||||||||||
| Balance at December 31, 2024 | $ | $ | $ | $ | |||||||||||||||||||
| Accruals | ( | ( | ( | ||||||||||||||||||||
| Cash payments | ( | ( | ( | ||||||||||||||||||||
| Balance at December 31, 2025 | $ | $ | $ | $ | |||||||||||||||||||
6. Inventories, net
Inventories, net consist of the following:
December 31, | |||||||||||
2025 | 2024 | ||||||||||
| Raw materials and supplies | $ | $ | |||||||||
| Work-in-process | |||||||||||
| Finished goods | |||||||||||
| Total inventories, net | $ | $ | |||||||||
105
7. Property, plant and equipment, net
Property, plant and equipment, net consists of the following:
December 31, | |||||||||||
2025 (1) | 2024 (1) | ||||||||||
| Land and improvements | $ | $ | |||||||||
| Buildings, building improvements and leasehold improvements | |||||||||||
| Furniture and equipment | |||||||||||
| Software | |||||||||||
| Construction-in-progress | |||||||||||
| Property, plant and equipment, gross | $ | $ | |||||||||
| Less: Accumulated depreciation and amortization | ( | ( | |||||||||
| Total property, plant and equipment, net | $ | $ | |||||||||
(1) During the years ended December 31, 2025 and 2024, the Company recorded non-cash impairment charges of $ | |||||||||||
For the years ended December 31, 2025 and 2024, construction-in-progress primarily included costs incurred to advance the Company's MCM Product capabilities.
8. Intangible assets and goodwill
Intangible Assets
The Company’s finite-lived intangible assets consist of products acquired via business combinations or asset acquisitions. The following table summarizes the Company’s finite-lived intangible assets:
December 31, 2025 | December 31, 2024 | ||||||||||||||||||||||||||||||||||||||||
Weighted Average Useful Life in Years | Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | Gross Carrying Amount | Accumulated Amortization | Net Carrying Amount | |||||||||||||||||||||||||||||||||||
Products | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||||||||||||||||
| Total intangible assets | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||||||||||||||||
Amortization expense associated with the Company’s finite-lived intangible assets was recorded as follows:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Amortization expense | $ | $ | $ | ||||||||||||||
106
The Company estimates our future amortization expense for our intangible assets as follows:
Year | As of December 31, 2025 | ||||
| 2026 | $ | ||||
| 2027 | |||||
| 2028 | |||||
| 2029 | |||||
| 2030 | |||||
| Thereafter | |||||
| Total remaining amortization | $ | ||||
Goodwill
The Company had no 218.2 million non-cash goodwill impairment charge for the MCM Products reporting unit, which is included in “Goodwill impairment” on the Consolidated Statement of Operations for the year ended December 31, 2023.
9. Fair value measurements
The table below presents information about the Company’s assets and liabilities that are regularly measured and carried at fair value and indicates the level within the fair value hierarchy of the valuation techniques the Company utilized to determine fair value:
December 31, 2025 | December 31, 2024 | ||||||||||||||||||||||||||||
Total | Level 1 | Level 2 | Level 3 | Total | Level 1 | Level 2 | Level 3 | ||||||||||||||||||||||
| Assets: | |||||||||||||||||||||||||||||
| Money market accounts | $ | $ | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||
| Total | $ | $ | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||
| Liabilities: | |||||||||||||||||||||||||||||
| Warrant liability | $ | $ | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||
| Total | $ | $ | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||
2024 Warrant liability
In connection with the Term Loan Agreement between the Company, OHA Agency LLC, as administrative agent, and the lenders from time to time party thereto (the “Term Loan Agreement”), the Company issued to the lenders warrants to purchase 1.0 million shares of the Company’s common stock at an exercise price of $9.8802 per share (the “Series I Warrants”) and warrants to purchase 1.5 million shares at an exercise price of $15.7185 per share (the “Series II Warrants” and, together with the Series I Warrants, the “Warrants”). The Warrants are currently exercisable and will expire on August 30, 2029. Because the Warrants could be cash settled based on events that are outside the control of the Company, it precludes the Warrants from applying the equity contract scope exception, and so are classified as a liability. As a result, the fair value of the Warrants will be remeasured each period with the gain or loss on the warrant liability included in “Other, net” on the Consolidated Statement of Operations. As of December 31, 2025 and December 31, 2024, the fair value of the warrant liability was $21.7 million and $16.2 million, respectively, and was included within “Other liabilities” on the Consolidated Balance Sheets, as determined using the Black-Scholes method.
107
The Company uses the Black-Scholes option pricing model to calculate the fair value of the Warrants at each reporting period. Assumptions used in the Black-Scholes option pricing model take into account the agreement terms as well as the quoted price of the Company’s common stock in an active market. The volatility is based on the average historical volatility of the common stock. The expected life is based on the remaining contractual term of the Warrants, and the risk free interest rate is based on the implied yield available on U.S. Treasury securities with a maturity equivalent to the Warrants’ expected life.
The table below is a reconciliation of the beginning and ending balance of the Company’s Level 3 warrant liability as of December 31, 2025 and 2024:
Warrant Liability | ||||||||
| Balance at December 31, 2023 | $ | |||||||
| Issuance of Warrants | ||||||||
| Change in fair value | ||||||||
| Balance at December 31, 2024 | $ | |||||||
| Change in fair value | ||||||||
| Balance at December 31, 2025 | $ | |||||||
The recurring Level 3 fair value measurement for the Company’s warrant liability used the following significant unobservable inputs:
Warrant Liability | Valuation Technique | Unobservable Input | Range | ||||||||
2024 Warrants | Black-Scholes Method | Term (in years) | |||||||||
| Risk free interest rate | |||||||||||
| Volatility | |||||||||||
Contingent consideration
Contingent consideration liabilities associated with business combinations required to be accounted for as derivatives are measured at fair value. The liabilities represent an obligation of the Company to transfer additional assets to the selling shareholders and owners if future events occur or conditions are met. These liabilities associated with business combinations are measured at fair value at inception and at each subsequent reporting date. The changes in the fair value are primarily due to the expected amount and timing of future net sales, which are inputs that have no observable market. Any change in fair value for the contingent consideration liabilities related to the Company’s products is classified on the Company's Consolidated Statements of Operations as “Cost of product and services sales, net.”
The following table is a reconciliation of the beginning and ending balance of the Company’s Level 3 contingent consideration liability measured at fair value as of December 31, 2024 and 2023. The Company had no Level 3 contingent consideration liabilities during 2025:
Contingent Consideration | |||||
| Balance at December 31, 2023 | $ | ||||
| Change in fair value | |||||
| Settlements | ( | ||||
Sales(1) | ( | ||||
| Balance at December 31, 2024 | $ | ||||
108
Non-variable rate debt
As of December 31, 2025 and 2024, the fair value of the Company's 3.875 389.7 million and $369.1 million, respectively. The fair value was determined through market sources, which are Level 2 inputs and directly observable. The carrying amounts of the Company’s other long-term variable interest rate debt arrangements approximate their fair values (see Note 11, “Debt” for more information regarding the Company’s repurchase of a portion of the Senior Unsecured Notes during the year ended December 31, 2025).
Non-recurring fair value measurements
10. Derivative instruments and hedging activities
Risk management objective of using derivatives
The Company is exposed to certain risks arising from both its business operations and economic conditions. The Company principally manages its exposures to a wide variety of business and operational risks through management of its core business activities. The Company manages economic risks, including interest rate, liquidity and credit risk primarily by managing the amount, sources and duration of its assets and liabilities and the use of derivative financial instruments. From time to time, the Company enters into interest rate swap transactions to manage exposures that arise from payments of variable interest rate debt associated with the Company's senior secured credit agreements. The objective and strategy with respect to these interest rate swaps is to protect the Company against adverse fluctuations in interest rates.
11. Debt
The table below present the components of the Company's debt:
December 31, | |||||||||||
2025 | 2024 | ||||||||||
| Senior secured credit agreement - Term loan due 2029 | $ | $ | |||||||||
| Total debt | $ | $ | |||||||||
| Unamortized debt issuance costs | ( | ( | |||||||||
| Non-current portion of debt | $ | $ | |||||||||
There were $35.0 million of unamortized debt issuance costs recorded in connection with the execution of the Term Loan Agreement within a contra account to directly offset the Term Loan balance. As of December 31, 2025, the Company has $15.1 million in unamortized debt issuance costs associated with the Term Loan.
Debt issuance costs associated with the Company’s Revolving Loans, as described in further detail below, were recorded as an asset within “Other long-term assets” on the Company’s Consolidated Balance Sheets. As of December 31, 2025, the Company has $2.7 million in unamortized debt issuance costs associated with the Revolving Loans. If the Company draws on the capacity available under the Revolving Loans, the debt issuance costs would be reclassified to a contra account to directly offset the Revolving Loans balance.
During the year ended December 31, 2024, the Company entered into a bilateral agreement with a bank in the amount of $5.1 million that is fully collateralized by cash, which is classified within “Restricted cash” in the Company’s Consolidated Balance Sheet as of December 31, 2024. As of December 31, 2025, $0.1 million remains in the "Restricted cash."
109
The Company recorded a gain on extinguishments of debt of $0.6 million during the year ended December 31, 2024 in “Gain (loss) on debt extinguishment” on the Consolidated Statements of Operations related to the Prior Credit Agreement (as defined below) and other loan forgiveness.
In August 2020, the Company issued $450.0 million aggregate principal amount of its Senior Unsecured Notes. Interest on the Senior Unsecured Notes is payable on February 15 and August 15 of each year until maturity, beginning on February 15, 2021. The 2028 Notes will mature on August 15, 2028.
The Company may redeem all or a portion of the Senior Unsecured Notes at a redemption price equal to 100 % of the principal amount of the Senior Unsecured Notes plus a “make-whole” premium and accrued and unpaid interest as set forth in the related indenture. Upon the occurrence of a change of control, the Company must offer to repurchase the Senior Unsecured Notes at a purchase price of 101 % of the principal amount of such notes plus accrued and unpaid interest.
Negative covenants in the indenture governing the 2028 Notes, among other things, limit the ability of the Company to incur indebtedness and liens, dispose of assets, make investments, enter into certain merger or consolidation transactions and make restricted payments.
In May 2025, the Board of Directors authorized the Company to repurchase up to $30.0 million in aggregate principal amount of the Company’s Senior Unsecured Notes. The Company may seek to opportunistically use this authority to repurchase its Senior Unsecured Notes in open market purchases, privately negotiated transactions or otherwise. Any such repurchases will depend upon prevailing market conditions, our liquidity requirements, contractual restrictions, applicable securities law and other factors. During the year ended December 31, 2025, the Company repurchased $10.3 million principal amount of its outstanding Senior Unsecured Notes. The Company paid $8.7 million in cash, including fees and accrued interest, in connection with the repurchases and recognized a gain on extinguishment of approximately $1.6 million for the year ended December 31, 2025, which was included in “Gain (loss) on debt extinguishment” on the Consolidated Statement of Operations. As of December 31, 2025, the Company had $19.7 million available to repurchase additional Senior Unsecured Notes.
Term Loan Agreement
On August 30, 2024, the Company entered into the Term Loan Agreement with OHA Agency LLC, as administrative agent, and the lenders from time to time party thereto. The Term Loan Agreement provides for a term loan (the “Term Loan”) of $250.0 million. The Term Loan was drawn in full on the date of entry into the Term Loan Agreement (the “Closing Date”). The Term Loan was issued with an original issue discount of 3.00 %.
The Term Loan will accrue interest at the Company’s option at (i) the Base Rate (as defined in the Term Loan Agreement) (subject to a floor of 1.00 %) plus 7.25 % per annum, referred to as “Term Base Rate Loans” or (ii) Adjusted Term Secured Overnight Financing Rate ("SOFR") (as defined in the Term Loan Agreement) (subject to a floor of 2.00 % until the second anniversary of the Closing Date, and thereafter, 3.00 %) plus 8.25 % per annum, referred to as “Term SOFR Loans”). A default interest rate of an additional 2.00 % per annum would apply on all outstanding obligations that are not paid when due. If any defaulted obligations are Term SOFR Loans, then such loans would, at the end of the applicable interest period, automatically be converted to Term Base Rate Loans that would continue to be subject to the default interest rate.
The Term Loan will mature on the first to occur (such date, the “Term Loan Maturity Date”) of (i) August 30, 2029, (ii) the date of acceleration of the Term Loan upon the occurrence and during the continuance of an event of default and (iii) solely to the extent the aggregate principal amount of Senior Unsecured Notes outstanding exceeds $25.0 million, May 15, 2028, which is three months prior to the August 15, 2028 maturity date of the Senior Unsecured Notes. The Term Loan Agreement contains certain customary default and cross-default provisions, representations and warranties and affirmative and negative covenants, including (a) restrictions on prepayments and repurchases of indebtedness, including the Senior Unsecured Notes, subject to further customary permitted debt payments (b) a minimum liquidity requirement of $75.0 million commencing on September 30, 2024 and tested every two weeks, and (c) a consolidated gross leverage ratio tested every fiscal quarter commencing with the fiscal quarter ending December 31, 2025, initially at 5.10 :1.00 with step-downs as set forth in the Term Loan Agreement. As of December 31, 2025, the Company was in compliance with all covenants under the Term Loan Agreement.
All indebtedness outstanding under the Term Loan Agreement is guaranteed by certain of the Company’s direct and indirect subsidiaries, other than certain subsidiaries that are not material, are excluded pursuant to the terms of the Term Loan Agreement, or will become guarantors on a post-closing basis (the Company and the guarantors, collectively, the Credit Parties”). The indebtedness under the Term Loan Agreement is secured by a first-priority security interest in and lien on substantially all assets of the Company and the other Credit Parties.
110
The Company may elect to prepay the Term Loan, in whole or in part, subject to (i) through and including the first anniversary of the Closing Date, a make-whole premium plus 4.00 % of the aggregate principal amount of the Term Loan subject to prepayment and (ii) after the first anniversary of the Closing Date, a 4.00 % prepayment premium, which percentage shall be reduced by 0.25 % as set forth on a schedule attached to the Term Loan Agreement. The Term Loan Agreement requires mandatory prepayments of the Term Loan in an amount equal to (a) 100 % of the aggregate net cash proceeds from the incurrence of certain indebtedness by the Term Loan Credit Parties and (b) (subject to certain reinvestment rights) 100 % of the aggregate net cash proceeds from (1) subject to certain specified exceptions, dispositions of property by the Credit Parties (provided that with respect to any dispositions occurring on or after the Closing Date, prepayment will not be required unless the net cash proceeds exceed $10.0 million in the aggregate per fiscal year or $5.0 million on a per-transaction basis) and (2) insurance proceeds received by any Credit Party or their subsidiaries resulting from theft, loss, physical destruction or damage of property. In December 2025, the Company voluntarily prepaid $100.0 million of the Term Loan, which was subject to the applicable $3.8 million prepayment premium under the Term Loan Agreement. The Company recognized a loss on extinguishment of $13.8 million, primarily attributable to the acceleration of unamortized debt issuance costs and prepayment premium, as recorded in “Gain (loss) on debt extinguishment” on the Consolidated Statement of Operations. As of December 31, 2025, the Company has $150.0 million in Term Loan principal remaining.
On the Closing Date, the Company used a portion of the proceeds of the Term Loan to repay all amounts outstanding and terminate commitments under the senior term loan facility under its Amended and Restated Credit Agreement, dated October 15, 2018, by and among the Company, the lenders party thereto from time to time, and Wells Fargo Bank, National Association, as the Administrative Agent (the “Prior Credit Agreement”), plus accrued interest and fees. The Company previously repaid all amounts outstanding under the revolving credit facility under the Prior Credit Agreement.
Revolving Loan Agreement
On September 30, 2024, the Company entered into the Revolving Credit Agreement with certain subsidiary borrowers (together with the Company, the “Borrowers”), the lenders from time to time party thereto, and Wells Fargo Bank, National Association, as agent (the “Agent”). The Credit Agreement provides for commitments with respect to the Revolving Loans (the "Revolving Loans") of up to the lesser of (x) $100.0 million, which may be increased (but not above $125.0 million, or the “Maximum Revolver Amount”) or decreased (but not below $50.0 million) by the Borrowers in accordance with the terms of the Revolving Credit Agreement and (y) the Borrowing Base (as defined in the Revolving Credit Agreement). Once reduced, the facility may not be increased. Up to $5.0 million of capacity under the Revolving Loans may be used for swing loans and up to $10.0 million may be used for the issuance of letters of credit.
The Revolving Loans will accrue interest at the Base Rate (as defined in the Revolving Credit Agreement) plus a margin of 1.25 % (such loans, “Revolving Base Rate Loans”) or, at the Company’s election, at a rate equal to Adjusted Term SOFR (as defined in the Revolving Credit Agreement and subject to a floor of 0.00 %) plus a margin of 2.25 % (such loans, “Revolving SOFR Loans”), in each case until September 30, 2025. After September 30, 2025, the applicable margin may be reduced to 0.75 % in the case of Revolving Base Rate Loans, or 1.75 % in the case of Revolving SOFR Loans, provided the Borrowers’ total leverage ratio is less than 4.00 to 1.00 for the most recently completed fiscal quarter and an event of default is not continuing. A default interest rate of an additional 2.00 % per annum would apply on all outstanding obligations that are not paid when due.
The Revolving Loans will mature on the first to occur of (i) September 30, 2029; (ii) to the extent there remain outstanding any portion of the term loans extended under the Term Loan Agreement, the date that is 90 days prior to the maturity date under the Term Loan Agreement; and (iii) to the extent any of the Senior Unsecured Notes remain outstanding, May 17, 2028, which is 90 days prior to the August 15, 2028 maturity date of the Senior Unsecured Notes. The Revolving Credit Agreement contains certain customary default and cross-default provisions (including with respect to defaults under the Term Loan Agreement), representations and warranties and affirmative and negative covenants, including (a) restrictions on prepayments and repurchases of indebtedness, including the Senior Unsecured Notes, (b) restrictions on dispositions of material intellectual property, (c) a minimum liquidity requirement of $50.0 million through the day prior to the first date following September 30, 2025 on which the Company’s total leverage ratio measured as of the preceding 12-month period is less than 5.25 to 1.00 (the “Covenant Conversion Date”) and (d) from the Covenant Conversion Date, a fixed charge coverage ratio requirement of at least 1.00 to 1.00. As of December 31, 2025, the Company was in compliance with all covenants under the Revolving Credit Agreement.
All indebtedness outstanding under the Revolving Credit Agreement is guaranteed by certain of the Borrowers’ material direct and indirect subsidiaries, subject to customary exclusions. The indebtedness under the Credit Agreement is secured by a first-priority security interest in and lien on the ABL Priority Collateral and a second-priority security interest and lien on the Term Loan Priority Collateral (in each case as defined in the Revolving Credit Agreement).
111
The Borrowers may elect to prepay any Revolving Loans, in whole or in part, without premium or penalty. If at any time outstanding Revolving Loans and letters of credit exceed the lesser of (i) the Borrowing Base, as adjusted for reserves established by the Agent, and (ii) the Maximum Revolver Amount, the Borrowers will be required to prepay outstanding obligations in the amount of such excess. The Agent may establish, increase or decrease reserves at its discretion.
Debt Maturity
Future debt payments of long-term indebtedness are as follows:
Year | As of December 31, 2025 | ||||
| 2026 | $ | ||||
| 2027 | |||||
| 2028 | |||||
| 2029 | |||||
| 2030 | |||||
| Thereafter | |||||
| Total debt | $ | ||||
12. Share-based compensation and stockholders' equity
Share-based compensation
The Company maintains the Emergent Plan and, until its termination on December 30, 2025, maintained the Inducement Plan. Both Plans include stock options, performance stock options, restricted stock units, performance stock units and liability classified long-term incentive awards.
As of December 31, 2025, an aggregate of 31.2 million shares of common stock were authorized for issuance under the Emergent Plan, of which a total of approximately 3.3 million shares of common stock remain available for future awards to be made to plan participants. As of December 31, 2025, an aggregate of 5.0 million shares of common stock had been authorized for issuance under the Inducement Plan and 1.1 million shares had been granted. The Inducement Plan was terminated on December 30, 2025, and the remaining 3.9 million authorized but unissued shares are no longer available for future awards. Additionally, during the year ended December 31, 2024, the Company granted an $8.0 million liability-classified long-term incentive award subject to performance based market conditions with the option to settle the award in any combination of cash or shares. The fair value of the liability-classified long-term incentive award was valued at grant using a Monte Carlo valuation model and will be revalued at each reporting period until the award is earned or expires. The long-term incentive award has a performance period of five years to vest based on the Company’s stock price performance.
Stock options and performance stock options
The Company utilizes the Black-Scholes valuation model for estimating the fair value of stock options granted. The exercise price of each option must be not less than 100 % of the fair market value of the shares underlying such option on the date of grant. Options granted under the Emergent Plan and the Inducement Plan have a contractual life of seven years . Set forth below are the assumptions used in valuing the stock options granted:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Expected dividend yield | % | % | % | ||||||||||||||
| Expected volatility | |||||||||||||||||
| Risk-free interest rate | |||||||||||||||||
Expected average life of stock options | |||||||||||||||||
112
During the year ended December 31, 2024, the Company granted performance stock options. These awards vest the later of one year after the grant date or the achievement of the market based performance condition. The performance period is five years after the grant date and the awards expire seven years after the grant date. The related stock-based compensation is recognized over the requisite service period, taking into account the probability that the market based performance condition will be achieved. The Company utilized the Monte Carlo valuation model for estimating the fair value of performance stock options granted. Set forth below are the assumptions used in valuing the performance stock options granted:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Expected dividend yield | |||||||||||||||||
| Expected volatility | % | ||||||||||||||||
| Risk-free interest rate | % | ||||||||||||||||
| Expected average life of performance stock options | — | — | |||||||||||||||
The following is a summary of stock option and performance stock option award activity under the Emergent Plan and the Inducement Plan:
Number of Shares | Weighted-Average Exercise Price | Weighted Average Remaining Contractual Term (in Years) | Aggregate Intrinsic Value | ||||||||||||||||||||
Stock options outstanding at December 31, 2024 | $ | ||||||||||||||||||||||
| Stock options granted | $ | ||||||||||||||||||||||
| Stock options exercised | ( | $ | |||||||||||||||||||||
| Stock options forfeited | ( | $ | |||||||||||||||||||||
| Stock options expired | ( | $ | |||||||||||||||||||||
Stock options outstanding at December 31, 2025 | $ | $ | $ | ||||||||||||||||||||
Stock options exercisable at December 31, 2025 | $ | $ | $ | ||||||||||||||||||||
There was $0.7 million, $0.3 million, and no cash received from option exercises for the years ended December 31, 2025, 2024 and 2023, respectively. There were no
The weighted average grant date fair value of options granted during the years ended December 31, 2025, 2024 and 2023 was $4.37 , $1.80 and $5.35 per share, respectively. The intrinsic value of stock options exercised is the amount by which the market value of our common stock on the exercise date exceeds the exercise price. The intrinsic value of options exercised during the year ended December 31, 2025 was $1.3 million due to a higher market price at the time of exercise relative to the strike price and higher option exercise activity. There was an immaterial intrinsic value of options exercised during the year ended December 31, 2024. There was no intrinsic value of options exercised during the year ended December 31, 2023.
The weighted average grant date fair value of performance options granted during the year ended December 31, 2024 was $1.13 . There were no
As of December 31, 2025, there was $7.1 million of unrecognized compensation cost related to stock options and $0.1 million of unrecognized compensation costs related to performance stock options. These costs are expected to be recognized over the weighted average periods of 1.5 years for stock options and 1.2 years for performance stock options.
113
Performance stock units and restricted stock units
The following is a summary of performance stock unit and restricted stock unit award activity under the Emergent Plan and the Inducement Plan:
Number of Shares | Weighted-Average Grant Date Fair Value | Aggregate Intrinsic Value | |||||||||||||||
Stock awards outstanding at December 31, 2024 | $ | $ | |||||||||||||||
| Stock awards granted | $ | ||||||||||||||||
| Stock awards released | ( | $ | |||||||||||||||
Stock awards forfeited (1) | ( | $ | |||||||||||||||
Stock awards outstanding at December 31, 2025 | $ | $ | |||||||||||||||
(1) Performance stock units forfeited during the year ended December 31, 2025 are included at the target payout percentage, or | |||||||||||||||||
The total fair value of restricted stock unit awards released during the years ended December 31, 2025, 2024 and 2023 was $9.4 million, $21.6 million and $31.8 million, respectively. As of December 31, 2025, there was $11.4 million of unrecognized compensation cost related to unvested restricted stock units. This cost is expected to be recognized straight-line over a weighted average period of 1.9 years.
Performance stock units represent common stock potentially issuable in the future, subject to achievement of performance conditions. Our current outstanding performance stock units vest based on certain financial metrics over the applicable performance period. The vesting and payout range for our performance stock units is typically between 50 % and up to 200 % of the target number of shares granted at the end of a or three-year performance period. There were no 2.4 million. As of December 31, 2025, there was $0.8 million of unrecognized compensation cost related to unvested performance stock units. That cost is expected to be recognized straight-line over a weighted average period of 1.2 years.
Share-based compensation expense
Share-based compensation expense, net of forfeitures was recorded in the following financial statement line items:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Cost of products and services sales, net | $ | $ | $ | ||||||||||||||
| R&D | |||||||||||||||||
| Selling, general and administrative | |||||||||||||||||
| Total share-based compensation expense | $ | $ | $ | ||||||||||||||
Stockholders' equity
Preferred stock
The Company is authorized to issue up to 15.0 million shares of preferred stock, $0.001 par value per share (“Preferred Stock”). Any Preferred Stock issued may have dividend rights, voting rights, conversion privileges, redemption characteristics, and sinking fund requirements as approved by the Company's board of directors.
Common stock
The Company currently has one class of common stock, $0.001 par value per share common stock (“Common Stock”), authorized and outstanding. The Company is authorized to issue up to 200.0 million shares of Common Stock. Holders of Common Stock are entitled to one vote for each share of Common Stock held on all matters, except as may be provided by law.
114
2025 Share Repurchase Program
In March 2025, the Company announced that its Board of Directors had authorized the repurchase of up to $50.0 million of the Company’s common stock (the “2025 Share Repurchase Program”) on or before March 27, 2026. In February 2026, the Company reauthorized the 2025 Share Repurchase Program for the repurchase of up to $50.0 million of the Company's common stock through March 31, 2027. Repurchases under the 2025 Share Repurchase Program may be made from time to time on the open market or in privately negotiated transactions. The timing and amount of any shares repurchased will be determined by the Company’s management based on its evaluation of market conditions and other factors, including the market price of the Company’s common stock, macroeconomic environment and other investment opportunities, consistent with applicable law. The 2025 Share Repurchase Program may be suspended or discontinued at any time. The Inflation Reduction Act of 2022 imposed a nondeductible 1% excise tax on the net value of certain stock repurchases made after December 31, 2022. Excise tax accrued during the year ended December 31, 2025 was $0.3 million.
During the year ended December 31, 2025, the Company utilized $25.1 million, including commissions and excise taxes, to repurchase 3.1 million shares. The average price paid, excluding commissions and excise taxes, was $8.15 per share. As of December 31, 2025, the Company had $24.9 million available to repurchase shares under the 2025 Share Repurchase Program.
2024 Issuance of Common Stock
In connection with the Term Loan Agreement, the Company entered into a Subscription Agreement, dated as of August 30, 2024 (the “Subscription Agreement”) with the lenders under the Term Loan Agreement, under which the Company issued 1.1 million shares of common stock with an aggregate value of $10.0 million in September 2024. At inception, the Subscription Agreement represented a forward sale of the Company’s common stock (the “Forward”). The Forward was initially classified and recorded as a liability and was remeasured to its fair value, resulting in a $1.6 million gain on settlement in September 2024.
2024 Warrant Issuance
In connection with the Term Loan Agreement, the Company issued to the lenders Series I Warrants to purchase 1.0 million shares of common stock and Series II Warrants to purchase 1.5 million shares of common stock. The Warrants are currently exercisable and will expire on August 30, 2029. Because the Warrants could be cash settled based on events that are outside the control of the Company, it precludes the Warrants from applying the equity contract scope exception, and so are classified as a liability. As of December 31, 2025, the fair value of the Warrants was $21.7 million. See Note 9, “Fair value measurements,” for more information on the accounting treatment and valuation of the Warrants.
As of December 31, 2025, the Company had the following Warrants outstanding to acquire shares of its common stock:
Warrants Outstanding | Range of Exercise Price per Share | Expiration Date | |||||||||
| Warrants issued related to the Term Loan Agreement | $ | August 2029 | |||||||||
| Total | |||||||||||
During the year ended December 31, 2025, no Warrants expired or were exercised.
At-the-Market Equity Offering Facility
The Company established an “at-the-market” equity offering program (the “ATM Program”) in May 2023. Pursuant to the ATM Program, the Company may, from time to time, sell up to $150.0 million aggregate gross sales price of shares of its common stock through Evercore Group L.L.C. and RBC Capital Markets, LLC, as sales agents. There were no 1.1 million shares of the Company’s common stock under the ATM Program for gross proceeds of $9.1 million, representing an average share price of $8.22 per share. As of December 31, 2025, $140.9 million aggregate gross sales price of shares of the Company’s common stock remains available for issuance under the ATM Program.
115
13. Earnings (loss) per common share
Basic earnings (loss) per common share is calculated using the treasury method by dividing net income (loss) by the weighted average number of shares of common stock outstanding during the period. Diluted earnings (loss) per common share adjusts basic earnings (loss) per common share for the effects of potentially dilutive common shares and is calculated using the treasury stock method. Potentially dilutive common shares include the dilutive effect of shares issuable under our equity compensation plans, including stock options, restricted stock units and performance stock units, as well as shares issuable upon exercises of the Warrants. Diluted earnings (loss) per share excludes anti-dilutive securities, which represent the number of potential common shares related to shares issuable under our equity compensation plans and pursuant to exercises of the Warrants that were excluded from diluted earnings (loss) per common share because their effect would have been antidilutive.
The following table presents the calculation of basic and diluted earnings (loss) per common share:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Numerator: | |||||||||||||||||
| Net income (loss) | $ | $ | ( | $ | ( | ||||||||||||
| Denominator: | |||||||||||||||||
| Weighted-average number of shares-basic | |||||||||||||||||
| Dilutive securities - equity awards | |||||||||||||||||
| Weighted-average number of shares-diluted | |||||||||||||||||
| Net income (loss) per common share - basic | $ | $ | ( | $ | ( | ||||||||||||
| Net income (loss) per common share - diluted | $ | $ | ( | $ | ( | ||||||||||||
The following table presents the securities and equity awards that are not considered in the diluted earnings (loss) per common share calculation generally because the exercise price of the awards was greater than the average per share closing price during the year ending December 31, 2025, 2024 and 2023 or the period resulted in a net loss, thus making the awards anti-dilutive. In certain instances, awards may be anti-dilutive even if the average market price exceeds the exercise price when the sum of the assumed proceeds exceeds the difference between the market price and the exercise price.
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Anti-dilutive stock awards | |||||||||||||||||
116
14. Revenue recognition
The Company's revenues disaggregated by the major sources were as follows:
Year Ended December 31, | |||||||||||||||||||||||||||||||||||||||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||||||||||||||||||||||||||||||||||||||
USG | Non-USG | Total | USG | Non-USG | Total | USG | Non-USG | Total | |||||||||||||||||||||||||||||||||||||||||||||
| Commercial Product sales | $ | $ | $ | $ | $ | $ | $ | $ | $ | ||||||||||||||||||||||||||||||||||||||||||||
| MCM Product sales | |||||||||||||||||||||||||||||||||||||||||||||||||||||
All other revenues (1) (2) | |||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total revenues | $ | $ | $ | $ | $ | $ | $ | $ | $ | ||||||||||||||||||||||||||||||||||||||||||||
(1) “All other revenues” includes Services and Contract and grants revenue. | |||||||||||||||||||||||||||||||||||||||||||||||||||||
(2) “All other revenues” for the year ended December 31, 2024 include $ | |||||||||||||||||||||||||||||||||||||||||||||||||||||
For the years ended December 31, 2025, 2024 and 2023, the Company's product sales from Naloxone products, Other Commercial products, Anthrax MCM, Smallpox MCM and Other products as a percentage of total product sales were as follows:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| % of product sales: | |||||||||||||||||
| Commercial Products: | |||||||||||||||||
| Naloxone products | % | % | % | ||||||||||||||
Other Commercial products (1) | % | % | % | ||||||||||||||
| MCM Products: | |||||||||||||||||
| Anthrax MCM | % | % | % | ||||||||||||||
| Smallpox MCM | % | % | % | ||||||||||||||
| Other Products | % | % | % | ||||||||||||||
(1) “Other Commercial products” refers to the travel health business that was sold to Bavarian Nordic in 2023. See Note 4, “Divestitures” for additional information related to the sale of the travel health business. | |||||||||||||||||
For the years ended December 31, 2025, 2024, and 2023, aside from sales to the USG, there were no sales to an individual customer in excess of 10% of total revenues. For the years ended December 31, 2025, 2024, and 2023, the Company’s revenues from customers within the United States comprised 71 %, 79 % and 58 %, respectively, of total revenues.
Transaction price allocated to remaining performance obligations
As of December 31, 2025, the Company has future contract value on unsatisfied performance obligations of approximately $244.4 million associated with all arrangements entered into by the Company. The Company expects to recognize $203.8 million of unsatisfied performance obligations within the next 24 months. The amount and timing of revenue recognition for unsatisfied performance obligations can change. The future revenues associated with unsatisfied performance obligations exclude the value of unexercised option periods in the Company’s revenue arrangements. Often the timing of manufacturing activities changes based on customer needs and resource availability. Government funding appropriations can impact the timing of product deliveries. The success of the Company's development activities that receive development funding support from the USG under development contracts can also impact the timing of revenue recognition.
Contract assets
The Company considers accounts receivable and deferred costs associated with revenue generating contracts, which are not included in inventory or property, plant and equipment and the Company does not currently have a contractual right to bill, to be contract assets. As of December 31, 2025 and December 31, 2024, the Company had $6.4 million and $9.7 million, respectively, of contract assets recorded within “Accounts receivable, net” on the Consolidated Balance Sheets.
117
Contract liabilities
When performance obligations are not transferred to a customer at the end of a reporting period, cash received associated with amounts allocated to those performance obligations is reflected as contract liabilities on the Consolidated Balance Sheets and is deferred until control of these performance obligations is transferred to the customer. The following table presents the roll forward of the contract liabilities:
Contract Liabilities | |||||
| Balance at December 31, 2024 | $ | ||||
| Balance at December 31, 2025 | $ | ||||
| Revenue recognized in the period from amounts included in contract liability at the beginning of the period: | $ | ||||
As of December 31, 2025 and 2024, the current portion of contract liabilities was $5.0 million and $4.8 million, respectively, and was included in “Other current liabilities” on the Consolidated Balance Sheet.
Accounts receivable and allowance for expected credit losses
Accounts receivable, including unbilled accounts receivable contract assets, consist of the following:
December 31, | |||||||||||
2025 | 2024 | ||||||||||
| Accounts receivable: | |||||||||||
| Billed | $ | $ | |||||||||
| Unbilled | |||||||||||
| Allowance for expected credit losses | ( | ( | |||||||||
| Accounts receivable, net | $ | $ | |||||||||
15. Leases
The Company is the lessee for operating leases for offices, R&D facilities and manufacturing facilities. The Company determines if an arrangement is a lease at inception. Operating leases are included in right-of-use assets and liabilities. The Company's leases have remaining lease terms of less than one year to approximately 9 years. Most leases included one or more options to renew, with renewal terms that can extend the lease term up to five years .
The components of lease expense were as follows:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Operating lease cost: | |||||||||||||||||
| Amortization of right-of-use assets | $ | $ | $ | ||||||||||||||
| Interest on lease liabilities | |||||||||||||||||
| Total operating lease cost | $ | $ | $ | ||||||||||||||
Operating lease costs are reflected as components of “Cost of products and services sales, net", “R&D” expense and “SG&A” expense on the Company's Consolidated Statements of Operations.
118
Supplemental balance sheet information related to leases was as follows:
December 31, | ||||||||||||||
Leases | Classification | 2025 | 2024 | |||||||||||
| Operating lease right-of-use assets | $ | $ | ||||||||||||
| Operating lease liabilities, current portion | $ | $ | ||||||||||||
| Operating lease liabilities | ||||||||||||||
| Total operating lease liabilities | $ | $ | ||||||||||||
| Operating leases: | ||||||||||||||
| Weighted average remaining lease term (years) | ||||||||||||||
| Weighted average discount rate | % | % | ||||||||||||
The maturity analysis below summarizes future undiscounted cash flows for our operating leases as of December 31, 2025:
Year | As of December 31, 2025 | ||||
| 2026 | |||||
| 2027 | |||||
| 2028 | |||||
| 2029 | |||||
| 2030 | |||||
| Thereafter | |||||
| Total undiscounted lease liabilities | |||||
| Less: Imputed interest | |||||
| Total lease liabilities | $ | ||||
16. Income taxes
The Company establishes valuation allowances for deferred income tax assets in accordance with U.S. GAAP, which provides that such valuation allowances shall be established unless realization of the income tax benefits is more likely than not. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible.
As of December 31, 2025, the Company reassessed the valuation allowance and considered negative evidence, including its significant losses in the current year and prior years, positive evidence, scheduled reversal of deferred tax liabilities, available taxes in carryback periods, tax planning strategies and projected future taxable income. After assessing both the negative and positive evidence, the Company concluded that it should record a net decrease in valuation allowance of $9.4 million on its global net operating losses, credits and other deferred tax assets.
The global intangible low-tax income (“GILTI”) provisions require the Company to include in its U.S. income tax return foreign subsidiary earnings in excess of an allowable return on the foreign subsidiary’s tangible assets. The Company is subject to incremental U.S. tax on GILTI income. The Company has elected to account for GILTI tax in the period in which it is incurred, and therefore has not provided any deferred tax amounts of GILTI in its consolidated financial statements for the years ended December 31, 2025, 2024 and 2023.
119
For the year ended December 31, 2025, the Company has re-evaluated its historical indefinite reinvestment assertion and determined it remains appropriate to record a deferred withholding tax liability for only the undistributed earnings of a certain subsidiary. The Company recognized a deferred withholding tax liability for the undistributed earnings of the Company’s international subsidiaries available cash and net working capital in the amount of $3.7 million. All other international subsidiaries’ outside basis differences are indefinitely reinvested.
Significant components of income taxes attributable to operations consist of the following:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Income (loss) from continuing operations before income tax expense (benefit): | |||||||||||||||||
| U.S. | $ | ( | $ | ( | $ | ( | |||||||||||
| International | |||||||||||||||||
| Income (loss) before income taxes | ( | ( | |||||||||||||||
| Income tax expense (benefit) from continuing operations: | |||||||||||||||||
| Current | |||||||||||||||||
| Federal | $ | $ | $ | ( | |||||||||||||
| State | |||||||||||||||||
| International | |||||||||||||||||
| Total current | |||||||||||||||||
| Deferred | |||||||||||||||||
| Federal | ( | ( | |||||||||||||||
| State | ( | ||||||||||||||||
| International | ( | ( | ( | ||||||||||||||
| Total deferred | ( | ( | ( | ||||||||||||||
Income tax provision | $ | $ | $ | ||||||||||||||
The table below presents the Company’s income taxes paid (net of refunds received) disaggregated by federal, state, and foreign jurisdictions:
December 31, | |||||
2025 | |||||
| Income taxes paid (refunded): | |||||
| U.S. Federal | $ | ||||
| U.S. State | |||||
| Foreign: | |||||
| Canada | |||||
| Other foreign | |||||
| Foreign Subtotal | |||||
| Total cash paid for income taxes (net of refunds) | $ | ||||
120
The Company adopted ASU 2023-09 prospectively; therefore, the current-year effective tax rate reconciliation below is presented in the new required format, while prior-year periods appear under the previous guidance. Income taxes differ from the amount of taxes determined by applying the U.S. federal statutory rate to income before taxes as a result of the following:
Year Ended December 31, 2025 | |||||||||||
Amount | Percent | ||||||||||
| Federal tax at statutory rates | $ | % | |||||||||
State taxes, net of federal benefit (1) | % | ||||||||||
| Foreign Tax Effects | |||||||||||
| Canada | |||||||||||
| Statutory tax rate difference between Canada and US | % | ||||||||||
| Local and Provincial taxes | % | ||||||||||
| Changes in valuation allowances | ( | ( | % | ||||||||
| Foreign Withholding Tax | % | ||||||||||
| Other | % | ||||||||||
| Ireland | |||||||||||
| Statutory tax rate difference between Ireland and US | ( | ( | % | ||||||||
| Effect of Cross Border Tax Laws | % | ||||||||||
| Effect of Cross-Border Tax Laws | ( | ( | % | ||||||||
| Changes in Valuation Allowances | % | ||||||||||
| Nontaxable or Nondeductible Items | |||||||||||
| Stock Compensation | % | ||||||||||
| Impact of Impairment | % | ||||||||||
| Compensation limitation | % | ||||||||||
| Debt Financing | % | ||||||||||
Impact of Divestitures | ( | ( | % | ||||||||
| Other | % | ||||||||||
| Effective Tax Rate | $ | % | |||||||||
(1) State taxes in California and Maryland made up the majority (greater than 50 percent) of the tax effect in this category. | |||||||||||
Year Ended December 31, | |||||||||||
2024 | 2023 | ||||||||||
| Federal tax at statutory rates | $ | ( | $ | ( | |||||||
| State taxes, net of federal benefit | ( | ( | |||||||||
| Impact of foreign operations | ( | ||||||||||
| Change in valuation allowance | |||||||||||
| Tax credits | ( | ( | |||||||||
| Pillar Two tax | |||||||||||
| Stock compensation | |||||||||||
| Goodwill Impairments | |||||||||||
| Adjustment of prior year taxes | |||||||||||
| Compensation limitation | |||||||||||
| Unrecognized tax benefit | ( | ( | |||||||||
| Impact of divestitures | ( | ||||||||||
| GILTI, net | |||||||||||
| Foreign withholding tax | |||||||||||
| Permanent differences | ( | ||||||||||
| Income tax provision | $ | $ | |||||||||
121
The effective annual tax rate for the years ended December 31, 2025, 2024, and 2023 was 36 %, (33 )% and (4 )%, respectively.
The effective annual tax rate of 36 % in 2025 is significantly different than the statutory rate primarily due to the impact of valuation allowance charge in the U.S., jurisdictional mix of income, and other permanent differences.
The effective annual tax rate of (33 )% in 2024 is significantly different than the statutory rate primarily due to the impact of reduced U.S. losses, valuation allowance charge in the U.S., jurisdictional mix of income, GILTI, and other permanent differences.
The effective annual tax rate of (4 )% in 2023 is significantly different than the statutory rate primarily due to the impact of a valuation allowance charge in the U.S., state and certain Foreign Jurisdictions, goodwill impairment, GILTI, and other permanent items. This is partially offset by tax credits and favorable rates in foreign jurisdictions.
The total unrecognized tax benefits recorded at December 31, 2025 and 2024 of $1.4 million and $1.7 million, respectively, is classified primarily as a non-current liability on the Consolidated Balance Sheets.
The table below presents the gross unrecognized tax benefits activity for the years ended December 31, 2025, 2024 and 2023:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Gross unrecognized tax benefits, beginning of period | $ | $ | $ | ||||||||||||||
| Increases (decreases) for tax positions for prior years | |||||||||||||||||
| Increases for tax positions for current year | |||||||||||||||||
| Settlements | ( | ||||||||||||||||
| Lapse of statute of limitations | ( | ( | ( | ||||||||||||||
| Gross unrecognized tax benefits, end of period | $ | $ | $ | ||||||||||||||
The current year change includes the reversal of a $0.3 million liability due to a lapse of the statute of limitations during the year.
The Company includes interest and potential penalties related to unrecognized tax benefits in income tax expense. As of December 31, 2025 and 2024, the total amount of interest and penalties accrued was $0.8 million and $0.8 million, respectively. The Company recognized interest and penalty expense (benefit) in 2025, 2024 and 2023 of $0.0 million, $(0.8 ) million and $0.2 million, respectively.
The Company does not anticipate a significant change within the next twelve months for unrecognized tax benefits and when resolved, all of these liabilities would impact the effective tax rate. However, the Company maintains a full valuation allowance as of December 31, 2025 and the recognition of any unrecognized tax benefits would be offset with a change in the valuation allowance and therefore there would be no income statement impact.
The Company's federal and state income tax returns for the tax years 2022 and onwards remain open to examination. The Company's tax returns for Canada remain open to examination for the tax years 2017 and onward. The Company's Irish tax returns remain open to examination for the tax years 2019 and onward.
As of December 31, 2025, the Company’s 2018 and 2020 Canadian Scientific Research and Experimental Development Claims are subject to proceedings with the Tax Court of Canada and the Company's 2021 Canadian Scientific Research and Experimental Development Claim is under audit. In addition, the Company’s 2021-2023 Texas Franchise tax returns are under audit.
122
The Company's net deferred tax liability consists of the following:
December 31, | |||||||||||
2025 | 2024 | ||||||||||
| Deferred tax assets | |||||||||||
| Federal losses carryforward | $ | $ | |||||||||
| State losses carryforward | |||||||||||
| R&D carryforward | |||||||||||
| Stock compensation | |||||||||||
| Foreign losses carryforward | |||||||||||
| Inventory reserves | |||||||||||
| Lease liability | |||||||||||
| IRC 263A capitalized costs | |||||||||||
| Capitalized R&D | |||||||||||
| IRC 163(j) Interest Limitation | |||||||||||
| Fixed assets | |||||||||||
| Intangible assets | |||||||||||
| Charitable Contributions | |||||||||||
| Reserves | |||||||||||
| Other | |||||||||||
| Gross deferred tax assets | |||||||||||
| Valuation allowance | ( | ( | |||||||||
| Total deferred tax assets | |||||||||||
| Deferred tax liabilities | |||||||||||
| Fixed assets | ( | ( | |||||||||
| Intangible assets | ( | ( | |||||||||
| Right-of-use asset | ( | ( | |||||||||
| Foreign Withholding Tax | ( | ( | |||||||||
| Prepaid expenses | ( | ( | |||||||||
| Other | ( | ( | |||||||||
| Total deferred tax liabilities | ( | ( | |||||||||
| Net deferred tax liabilities | $ | ( | $ | ( | |||||||
As of December 31, 2025, the Company has approximately $547.2 million in U.S. federal net operating loss ("NOL") carryforwards, $36.0 million of NOL’s which will expire in varying amounts in 2031 through 2035 and $511.2 million which will carryforward indefinitely, although, limited to eighty percent of taxable income annually. The Company has U.S. federal R&D tax credit carryforwards of $17.8 million which will expire in 2027 through 2042.
As of December 31, 2025, the Company had post-apportionment state NOLs totaling approximately $1.1 billion that will begin to expire in 2028. The Company has state R&D tax credit carryforwards of $5.0 million which will expire in 2027 through 2038.
The deductibility of such U.S. federal and state net operating losses and credits may be limited. Under Section 382/383 of the Internal Revenue Code of 1986, as amended (the “Code”), and corresponding provisions of state law, if a corporation undergoes an "ownership change," which generally occurs if the percentage of the corporation's stock owned by 5% stockholders increases by more than 50% over a three-year period, the corporation's ability to use its pre-change NOL carryforwards and other pre-change tax attributes to offset its post-change income may be limited. Certain of the net operating loss carryforwards and the credit carryforwards are subject to an annual limitation pursuant to Code Section 382 and 383 as a result of historical acquisitions. We may experience ownership changes in the future as a result of subsequent shifts in our stock ownership, some of which may be outside of our control, which may further limit our carryforwards. If we determine that an ownership change has occurred and our ability to use our historical NOL and credit carryforwards is materially limited, it would harm our future operating results by effectively increasing our future tax obligations.
123
The Company has approximately $58.7 million in net operating losses from foreign jurisdictions as of December 31, 2025, which will carryforward indefinitely.
The Company’s valuation allowance decreased by $9.4 million due to the Company’s release of Canadian valuation allowance and generation of significantly lower losses in 2025.
17. Defined benefit and 401(k) savings plan
Defined benefit pension plan
The Company previously sponsored a defined benefit pension plan covering eligible employees in Switzerland (the “Swiss Plan”), which was sold as part of the divestiture of the Company's travel health business to Bavarian Nordic in 2023 (See Note 4, “Divestitures” for information on the sale of the travel health business). The Company recognized no 0.6 million, reflected as a component of “Selling, general and administrative” expenses on the Consolidated Statement of Operations.
401(k) savings plan
The Company has established a defined contribution savings plan under Section 401(k) of the Internal Revenue Code (the “401(k) Plan”). The 401(k) Plan covers substantially all U.S. employees. Under the 401(k) Plan, employees may make elective salary deferrals. During the years ended December 31, 2025, 2024 and 2023, the Company made matching contributions of approximately $4.5 million, $5.7 million and $8.3 million, respectively.
18. Purchase commitments
The Company enters into agreements in the normal course of business with vendors for raw materials and other goods or services. Purchase commitments are agreements to purchase raw materials and services that are enforceable, legally binding, and specify terms that (1) include fixed or minimum quantities to be purchased, (2) include fixed, minimum or variable price provisions and (3) are longer than one year. Purchase commitments exclude agreements that are cancellable without penalty.
As of December 31, 2025, the Company has approximately $474.8 million of purchase commitments associated with raw materials and Bioservices that will be purchased in the next five years , of which the Company estimates that approximately $142.9 million will be purchased within the next year. For the years ended December 31, 2025, 2024, and 2023, the Company purchased $88.6 million, $96.1 million and $107.8 million, respectively, of materials and services under these commitments.
124
19. Segment information
In the first quarter of 2025, using the guidance provided in ASC 280, Segment Reporting (“ASC 280”), along with the adoption of ASU 2023-07, Segment Reporting (Topic 280), the Company reevaluated its reportable and operating segments. Based on updates to the Company’s internal operating and reporting structure and quantitative tests outlined in ASC 280, the Company manages its business with a focus on two reportable segments; the Commercial Products segment, which includes Naloxone products and the MCM Products segment, which includes the Anthrax - MCM products, Smallpox - MCM products and Other Products. The Company’s Services operating segment no longer meets the quantitative threshold for determining reportable segments and is now included within “All other revenues” along with the Company’s Contracts and grants business.
The Company's Chief Operating Decision Maker (“CODM”) is its President and Chief Executive Officer. The CODM evaluates the performance of the Company's reportable segments based on segment adjusted gross margin. The Company defines segment adjusted gross margin as sales less cost of sales excluding restructuring costs (benefits), changes in fair value of financial instruments, and inventory step-up provision for each reportable segment. The Company does not allocate amortization of intangible assets, research and development expenses, selling, general and administrative costs, interest and other income (expense) or taxes to each reportable segment in the operating results that are regularly reviewed by the CODM. The CODM uses these reported measures to assess segment performance, allocate resources and monitor budget and guidance versus actual results. These metrics are used by the CODM to make key operating decisions, such as decisions about allocating capital and other resources to each segment. The accounting policies for segment reporting are the same as those described in Note 2, “Summary of significant accounting policies”. Intersegment revenue, cost of sales, and profit are eliminated in the segment measures regularly reviewed by the CODM as these activities are eliminated in consolidation and thus are not included in management’s evaluation of performance for each segment.
The Company manages its assets on a total company basis, not by segment, as the Company's assets are shared or commingled. Therefore, the Company’s CODM does not regularly review any asset information by segment and, accordingly, the Company does not report asset information by segment. The measure of segment assets is reported on the Consolidated Balance Sheet as “Total assets”.
For all tables presented below, the prior period disclosures have been recast to conform to the current period segment presentation.
125
The following table presents segment information provided to the CODM, along with a reconciliation of segment adjusted gross margin to loss before income taxes as reported in the Consolidated Statement of Operation for the years ended December 31, 2025, 2024, and 2023:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Revenues: | |||||||||||||||||
Commercial Products | $ | $ | $ | ||||||||||||||
| MCM Products | |||||||||||||||||
Reconciliation of revenue: | |||||||||||||||||
All other revenues (1) | |||||||||||||||||
| Total revenues | $ | $ | $ | ||||||||||||||
| Cost of sales reportable segments: | |||||||||||||||||
Commercial Products (2) | $ | $ | $ | ||||||||||||||
MCM Product (3) | |||||||||||||||||
| Total of reportable segments | |||||||||||||||||
| Segment adjusted gross margin reportable segments: | |||||||||||||||||
| Commercial Products | $ | $ | $ | ||||||||||||||
| MCM Products | |||||||||||||||||
| Total of reportable segments | $ | $ | $ | ||||||||||||||
| Reconciliation to income (loss) before income tax: | |||||||||||||||||
All other revenues less other costs of revenue (1) | $ | $ | ( | $ | ( | ||||||||||||
| Amortization of intangible assets | ( | ( | ( | ||||||||||||||
Restructuring benefits (costs) | ( | ( | |||||||||||||||
Inventory step-up provision | ( | ( | ( | ||||||||||||||
Changes in fair value of financial instruments | ( | ( | |||||||||||||||
Settlement charge, net | ( | ||||||||||||||||
| Goodwill impairment | ( | ||||||||||||||||
| Impairment of long-lived assets | ( | ( | ( | ||||||||||||||
| Research and development | ( | ( | ( | ||||||||||||||
| Selling, general and administrative | ( | ( | ( | ||||||||||||||
| Interest expense | ( | ( | ( | ||||||||||||||
| Gain on sale of business | |||||||||||||||||
| Gain (loss) on debt extinguishment | ( | ||||||||||||||||
| Other, net | |||||||||||||||||
| Income (loss) before income taxes | $ | $ | ( | $ | ( | ||||||||||||
(1) "All other revenues" and "All other revenue less other cost of revenue" include Services and Contracts and grants revenue, and Services and Contracts and grants revenue less Cost of services, respectively. | |||||||||||||||||
(2) Excludes $ | |||||||||||||||||
(3) Excludes $ | |||||||||||||||||
126
The following table includes depreciation expense for each reportable segment:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Depreciation from reportable segments: | |||||||||||||||||
| Commercial Products | $ | $ | $ | ||||||||||||||
| MCM Products | |||||||||||||||||
| Items not included in depreciation from reportable segments: | |||||||||||||||||
| All other segment | |||||||||||||||||
| Other | |||||||||||||||||
| Total | $ | $ | $ | ||||||||||||||
The following table includes revenues by country. Revenues have been attributed based on the location of the customer:
Year Ended December 31, | |||||||||||||||||
2025 | 2024 | 2023 | |||||||||||||||
| Revenue: | |||||||||||||||||
| United States | $ | $ | $ | ||||||||||||||
| Canada | |||||||||||||||||
| Other | |||||||||||||||||
| Total revenues | $ | $ | $ | ||||||||||||||
The following table includes long-lived assets, net by country. Long-lived assets, net includes right-of-use assets and property, plant & equipment, net, excluding software, net:
December 31, | |||||||||||
2025 | 2024 | ||||||||||
| Long-lived assets, net: | |||||||||||
| United States | $ | $ | |||||||||
| Canada | |||||||||||
| Other | |||||||||||
| Total Long-lived assets, net | $ | $ | |||||||||
20. Litigation
Securities and shareholder litigation
With respect to the specific legal proceedings and claims described below, unless otherwise noted, the amount or range of possible losses is not reasonably estimable. There can be no assurance that the settlement, resolution, or other outcome of one or more matters, including the matters set forth below, during any subsequent reporting period will not have a material adverse effect on the Company's results of operations or cash flows for that period or on the Company's financial condition.
Between April and June 2021 three putative class action lawsuits were filed against the Company and certain of its current and former senior officers in the United States District Court for the District of Maryland (the "Court") on behalf of purchasers of the Company’s common stock, seeking to pursue remedies under the Exchange Act. These cases were consolidated in December 2021 under the caption In re Emergent BioSolutions Inc. Securities Litigation, No. 8:21-cv-00955-PWG (the "Federal Securities Class Action").
In September 2024, the Company and the lead plaintiffs in the consolidated matter entered into an agreement in principle to settle the claims against the Company and each of the Company’s current and former officers and directors. In October 2024, the Court granted preliminary approval of the proposed settlement, ordered notice to the settlement class and scheduled a fairness hearing that was held in February 2025. At the scheduled fairness hearing in February 2025, the Court granted final approval of the settlement. Under the settlement, the claims against the Company and its officers and directors were dismissed with prejudice and released in exchange for a payment from the Company of $40.0 million, $30.0 million of which was paid from insurance proceeds, and had previously been funded in the fourth quarter of 2024. The Company recorded the settlement and insurance recoverable amounts as pre-tax operating expense and income, respectively, within “Selling, general and administrative” expenses on the Consolidated Statement of Operations for the year ended December 31, 2024.
127
In June 2021, Lincolnshire Police Pension Fund (“Lincolnshire”), and in August 2021, Pooja Sayal, filed putative shareholder derivative lawsuits in the United States District Court for the District of Maryland on behalf of the Company against certain of the Company's current and former officers and directors for breach of fiduciary duties, waste of corporate assets, and unjust enrichment, each allegation related to the CDMO Manufacturing Capabilities. In addition to monetary damages, the complaints sought the implementation of multiple corporate governance and internal policy changes. In November 2021, the cases were consolidated under the caption In re Emergent BioSolutions Inc. Stockholder Derivative Litigation, Master Case No. 8:21-cv-01595-DLB. In January 2022, the Lincolnshire complaint was designated as the operative complaint in the consolidated action. In April 2022, the Court approved the parties' joint stipulation to and stay of the proceedings and discovery until the close of fact discovery in the Federal Securities Class Action.
In September 2021 and November 2021, three putative shareholder derivative lawsuits were filed by Chang Kyum Kim, Mark Nevins and Employees Retirement System of the State of Rhode Island, North Collier Fire Control and Rescue District Firefighters Pension Plan, and Pembroke Pines Firefighters & Police Officers Pension Fund, respectively, in the Court of Chancery of the State of Delaware on behalf of the Company against certain of its current and former officers and directors for breach of fiduciary duties, unjust enrichment and insider trading, each allegation related to the CDMO Manufacturing Capabilities. In addition to monetary damages, the complaints sought the implementation of multiple corporate governance and internal policy changes. In February 2022, the cases were consolidated under the caption In re Emergent BioSolutions, Inc. Derivative Litigation, C.A. No. 2021-0974-MTZ with the institutional investors as co-lead plaintiffs. On March 4, 2022, the defendants filed a motion to dismiss the complaint. On March 29, 2022, an order was granted staying all proceedings pending a final, non-appealable judgment in the Federal Securities Class Action.
In December 2021 and January 2022, three putative shareholder derivative lawsuits were filed by Zachary Elton, Eric White and Jeffrey Reynolds, respectively, in the Circuit Court for Montgomery County, Maryland on behalf of the Company against certain of its current and former officers and directors for breach of fiduciary duty, unjust enrichment, waste of corporate assets, failing to maintain internal controls, making or causing to be made false and/or misleading statements and material omissions, insider trading and otherwise violating the federal securities laws, each allegation related to the CDMO Manufacturing Capabilities. The complaints sought monetary and punitive damages. In February 2022, the Court entered an order consolidating these actions under case number C-15-21-CV-000496. On March 9, 2022, the parties filed a Joint Stipulation of Stay of Proceedings and Discovery, pursuant to which the parties agreed to stay all proceedings until 30 calendar days after a ruling on the defendants’ motion to dismiss, and in November 2023, the Court approved the parties' joint stipulation to extend the stay of the proceedings and discovery until the close of fact discovery in the Federal Securities Class Action.
In March 2025, plaintiffs Lincolnshire and Pooja Sayal filed a motion in the United States District Court for the District of Maryland seeking preliminary approval of a stipulation of settlement with regard to the above referenced derivative matters (the “Proposed Settlement”). The Proposed Settlement provided that the defendants must cause their insurers to pay to the Company a settlement amount of $15.0 million, less a court-approved fee and expense amount (the “Settlement Amount”) and implement certain governance reforms. In August 2025, the Court granted approval of the Proposed Settlement without modification. The Company received the Settlement Amount in September 2025. Accordingly, during the year ended December 31, 2025, the Company recorded $10.5 million with respect to the Settlement Amount as a reduction of “Selling, general and administrative” expenses on the Consolidated Statement of Operations.
Government investigations
In the second quarter of 2025, the Company met with representatives of the Department of Justice regarding the Company’s 2017 and 2019 contracts with the Department of State to provide medical countermeasures for exposure to nerve toxins. The Company has provided documents and is continuing discussions with the Department of Justice on this matter.
In the first quarter of 2025, the Company received an inquiry from the New York Attorney General’s Office (OAG) related to certain past trading activity by the Company’s former Chief Executive Officer. In January 2026, the Company announced that it had reached an agreement with the OAG to resolve this investigation. Under the terms of the agreement, the Company agreed to pay $0.9 million to the State of New York and to implement enhancements to its Insider Trading Policy. The $0.9 million was recorded within “Selling, general and administrative” expenses on the Consolidated Statement of Operations for the year ended December 31, 2025 and paid subsequent to year end. In addition, the Company agreed to provide reporting to the OAG for a period of three years regarding trading plans adopted, modified, or terminated by senior management and board members.
128
In the second quarter of 2021, the Company received subpoenas from the SEC related to certain disclosures regarding the incidents described above under the heading “Securities and shareholder litigation.” The Company cooperated with the SEC’s investigation. During the first quarter of 2025, the Company determined that a loss resulting from the investigation was probable and that the amount of loss could be reasonably estimated. As a result, the Company recorded an accrual within “Selling, general and administrative” expenses on the Consolidated Statement of Operations for the year ended December 31, 2024 of $1.5 1.5 million. The Company paid the fine on April 18, 2025 and no liability balance remains on the Company’s Consolidated Balance Sheet as of December 31, 2025.
2022 Termination of manufacturing services agreement with Janssen Pharmaceuticals, Inc.
In July 2020, the Company, through its wholly owned subsidiary, Emergent Manufacturing Operations Baltimore, LLC, entered into the Janssen Agreement with Janssen for large-scale drug substance manufacturing of Johnson & Johnson’s investigational SARS-CoV-2 vaccine, Ad26.COV2-S, recombinant based on the AdVac technology (the “Product”).
In June 2022, the parties exchanged notices alleging material breaches of the Janssen Agreement, with each asserting the other had failed to meet key contractual obligations. Janssen subsequently initiated arbitration proceedings, and the Company responded with counterclaims, which were resolved by Settlement Agreement in July 2024. Pursuant to the terms of the Settlement Agreement, Janssen paid the Company $50.0 million on July 31, 2024.
In 2022, because the arbitration process with Janssen was expected to extend longer than one year, the Company reclassified amounts related to the Janssen Agreement from “Inventories, net” and from “Prepaid expenses and other current assets” to “Other assets”, resulting in $152.7 million in long-term assets related to the Janssen Agreement on the Consolidated Balance Sheet as of December 31, 2022. The long-term asset balance within “Other Assets” prior to announcing the Settlement Agreement was $158.7 million. The Company recorded $50.0 110.2 million in the second quarter of 2024 within “Cost product and services sales, net” on the Consolidated Statement of Operations to write down the remaining inventory to its net realizable value and for estimated disposal costs. The receivable for the settlement amount was collected during the third quarter of 2024 and there was no long-term asset balance remaining within “Other Assets” related to the Janssen Agreement as of December 31, 2024.
129
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Not applicable.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our chief executive officer and chief financial officer, evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2025. The term "disclosure controls and procedures," as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC's rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company's management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2025, our chief executive officer and chief financial officer concluded that, as of such date, that the disclosure controls and procedures were effective at the reasonable assurance level.
Management's Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our system of internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements. Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2025. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013 Framework). As a result of this assessment, our management concluded that, as of December 31, 2025, our internal control over financial reporting was effective based on those criteria.
Ernst & Young LLP, the independent registered public accounting firm that has audited our consolidated financial statements included herein, has issued an attestation report on the effectiveness of our internal control over financial reporting as of December 31, 2025, a copy of which is included in this Annual Report on Form 10-K.
Changes in Internal Control Over Financial Reporting
There have been no changes in the Company's internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) that occurred during the quarter ended December 31, 2025 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
130
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Emergent BioSolutions Inc.
Opinion on Internal Control over Financial Reporting
We have audited Emergent BioSolutions Inc. and subsidiaries’ internal control over financial reporting as of December 31, 2025, based on criteria established in Internal Control — Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Emergent BioSolutions Inc. and subsidiaries (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2025, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 2025 and 2024, the related consolidated statements of operations, comprehensive income (loss), changes in stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2025, and the related notes and financial statement schedule listed in the Index at Item 15 and our report dated February 26, 2026, expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Tysons, Virginia
February 26, 2026
131
ITEM 9B. OTHER INFORMATION
During the three months ended December 31, 2025, directors and officers (as defined in Exchange Act Rule 16a-1(f)) of the Company adopted written plans for the sale of the Company’s common stock intended to satisfy the affirmative defense conditions of Exchange Act Rule 10b5-1(c) (“Rule 10b5-1 trading arrangements”) as set forth in the table below.
Name | Position | Action | Adoption date | Rule 10b5-1 Trading Plan Provides for Purchase/Sale (1) | Total Shares of Common Stock to be Sold | Expiration Date (2) | ||||||||||||||
Sale | ||||||||||||||||||||
Sale | ||||||||||||||||||||
Sale | ||||||||||||||||||||
Each of the above-named directors and officers is currently and is expected to remain in compliance with his or her share ownership guidelines following the sale of any common shares pursuant to his or her Rule 10b5-1 trading arrangement
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS.
Not applicable.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Code of Ethics
We have adopted a code of business conduct and ethics that applies to our directors, officers (including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions), as well as our other employees. A copy of our code of business conduct and ethics is available on our website at www.emergentbiosolutions.com. We intend to post on our website all disclosures that are required by applicable law, the rules of the SEC or the New York Stock Exchange concerning any amendment to, or waiver of, our code of business conduct and ethics. The reference to our website is intended to be an inactive textual reference only. Neither the information on nor that can be accessed through our website are incorporated by reference in this Annual Report on Form 10-K.
The remaining information required by Item 10 is hereby incorporated by reference from our Definitive Proxy Statement relating to our 2026 Annual Meeting of Stockholders, which we expect to file with the U.S. Securities and Exchange Commission (“SEC”) within 120 days following the end of our fiscal year.
ITEM 11. EXECUTIVE COMPENSATION
The information required by Item 11 is hereby incorporated by reference from our Definitive Proxy Statement relating to our 2026 Annual Meeting of Stockholders, which we expect to file with the SEC within 120 days following the end of our fiscal year.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by Item 12 is hereby incorporated by reference from our Definitive Proxy Statement relating to our 2026 Annual Meeting of Stockholders, which we expect to file with the SEC within 120 days following the end of our fiscal year.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by Item 13 is hereby incorporated by reference from our Definitive Proxy Statement relating to our 2026 Annual Meeting of Stockholders, which we expect to file with the SEC within 120 days following the end of our fiscal year.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required by Item 14 is hereby incorporated by reference from our Definitive Proxy Statement relating to our 2026 Annual Meeting of Stockholders, which we expect to file with the SEC within 120 days following the end of our fiscal year.
132
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
Financial Statements
The following financial statements and supplementary data are filed as a part of this Annual Report on Form 10-K in Part II, Item 8.
•Report of Independent Registered Public Accounting Firm (PCAOB ID: 42 )
•Consolidated Balance Sheets at December 31, 2025 and 2024
•Consolidated Statements of Operations for the years ended December 31, 2025, 2024 and 2023
•Consolidated Statements of Comprehensive Income (Loss) for the years ended December 31, 2025, 2024 and 2023
•Consolidated Statements of Cash Flows for the years ended December 31, 2025, 2024 and 2023
•Consolidated Statement of Changes in Stockholders' Equity for the years ended December 31, 2025, 2024 and 2023
•Notes to Consolidated Financial Statements
Financial Statement Schedules
Schedule II - Valuation and Qualifying Accounts for the years ended December 31, 2025, 2024 and 2023 has been filed as part of this annual report on Form 10-K. All other financial statement schedules are omitted because they are not applicable or the required information is included in the financial statements or notes thereto.
Exhibits
Those exhibits required to be filed by Item 601 of Regulation S-K are listed in the Exhibit Index immediately preceding the exhibits hereto and such listing is incorporated herein by reference.
SCHEDULE II - VALUATION AND QUALIFYING ACCOUNTS
(in millions) | Beginning Balance | Charged to Costs and Expenses | Deductions | Ending Balance | ||||||||||||||||||||||
| Year Ended December 31, 2025 | ||||||||||||||||||||||||||
| Prepaid expenses and other current assets allowance | $ | ( | $ | |||||||||||||||||||||||
| Year Ended December 31, 2024 | ||||||||||||||||||||||||||
| Prepaid expenses and other current assets allowance | $ | ( | $ | |||||||||||||||||||||||
| Year Ended December 31, 2023 | ||||||||||||||||||||||||||
| Prepaid expenses and other current assets allowance | $ | ( | $ | |||||||||||||||||||||||
133
Exhibit Index
All documents referenced below were filed pursuant to the Securities Exchange Act of 1934 by the Company, (File No. 001-33137), unless otherwise indicated.
Exhibit Number | Exhibit Description | |||||||
| 3.1 | Third Restated Certificate of Incorporation of the Company (incorporated by reference to Exhibit 3 to the Company's Quarterly Report on Form 10-Q filed on August 5, 2016). | |||||||
| 3.2 | Amended and Restated By-laws of the Company (incorporated by reference to Exhibit 3 to the Company's Current Report on Form 8-K filed on August 16, 2012). | |||||||
| 4.1 | Description of the Company's Securities (incorporated by reference to Exhibit 4.1 to the Company’s Annual Report on Form 10-K, filed on March 4, 2025). | |||||||
| 4.2 | Specimen Common Stock Certificate (incorporated by reference to Exhibit 4.1 to Amendment No. 3 to the Company's Registration Statement on Form S-1 filed on October 20, 2006) (Registration No. 333-136622). | |||||||
| 4.3 | ||||||||
| 4.4 | First Supplemental Indenture, dated as of August 30, 2024, by and among Emergent BioSolutions Inc., the subsidiary guarantors party thereto and U.S. Bank Trust Company, National Association, as trustee (incorporated by reference to Exhibit 4.2 to the Company’s Current Report on Form 8-K, filed on September 3, 2024). | |||||||
| 4.5 | Form of 3.875% Senior Unsecured Note due 2028 (incorporated by reference to Exhibit 4.1 to the Company's Current Report on Form 8-K, filed on August 7, 2020.) (incorporated by reference to Exhibit 4.2 to the Company's Quarterly Report on Form 10-Q filed on November 6, 2020). | |||||||
| 4.6 | Form of Warrant (included in Exhibit 10.4). | |||||||
| 10.1 | Equity Distribution Agreement, dated May 17, 2023 (incorporated by reference to Exhibit 10.2 to the 8-K filed on May 18, 2023). | |||||||
| 10.2 | †† | Credit Agreement, dated as of August 30, 2024, by and among Emergent BioSolutions Inc., the lenders from time to time party thereto and OHA Agency LLC, as administrative agent (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed on September 3, 2024). | ||||||
| 10.3 | †† | Credit Agreement, dated as of September 30, 2024, by and among Emergent BioSolutions Inc., certain of its subsidiaries, as borrowers, the lenders party thereto from time to time, and Wells Fargo Bank, National Association, as the agent (incorporated by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K, filed on October 2, 2024). | ||||||
| 10.4 | Warrant Agreement between Emergent BioSolutions Inc. and Broadridge Corporate Issuer Solutions LLC, as Warrant Agent, dated August 30, 2024 (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K, filed on September 3, 2024). | |||||||
| 10.5 | * | Emergent BioSolutions Inc. Amended and Restated Stock Incentive Plan (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K, filed on May 29, 2024). | ||||||
| 10.6 | * | Emergent BioSolutions Inc. Amended Employee Stock Purchase Plan (incorporated by reference to Exhibit 4.5 to the Registration Statement on Form S-8, filed on June 5, 2023). | ||||||
| 10.7 | * | Executive Agreement, dated February 19, 2024 between Emergent BioSolutions Inc. and Joseph Papa (incorporated by reference to Exhibit 10 to the Company's Current Report on Form 8-K filed on February 21, 2024). | ||||||
| 10.8 | * | Form of Director Nonstatutory Stock Option Agreement (incorporated by reference to Exhibit 10.10 to the Company’s Annual Report on Form 10-K filed on February 22, 2019). | ||||||
| 10.9 | * | Form of Director Restricted Stock Unit Agreement (incorporated by reference to Exhibit 10.11 to the Company’s Annual Report on Form 10-K filed on February 22, 2019). | ||||||
| 10.10 | * | Global Form of Restricted Stock Unit Award Agreement (incorporated by reference to Exhibit 10.13 to the Company's Annual Report on Form 10-K filed on February 19, 2021). | ||||||
| 10.11 | * | Global Form of Non-Qualified Stock Option Agreement (incorporated by reference to Exhibit 10.11 to the Company’s Annual Report on Form 10-K filed on February 25, 2020). | ||||||
| 10.12 | ††* | Form of 2023-2025 Performance Based Stock Unit Award Agreement (incorporated by reference to Exhibit 10 to Current Report on Form 8-K filed on March 23, 2023). | ||||||
| 10.13 | * | Form of Indemnity Agreement for Directors and Senior Officers (incorporated by reference to Exhibit 10 to the Company's Current Report on Form 8-K filed on January 18, 2013). | ||||||
| 10.14 | * | Annual Bonus Plan for Executive Officers (incorporated by reference to Exhibit 10.7 to the Company's Annual Report on Form 10-K filed on March 5, 2010). | ||||||
134
Exhibit Number | Exhibit Description | |||||||
| 10.15 | * | Second Amended and Restated Senior Management Severance Plan (incorporated by reference to Exhibit 10 to the Company's Current Report on Form 8-K filed on July 16, 2015). | ||||||
| 10.16 | † | Solicitation/Contract/Order for Commercial Items (the CDC BioThrax Procurement Contract), effective December 8, 2016, from the Centers for Disease Control and Prevention to Emergent Biodefense Operations Lansing LLC (incorporated by reference to Exhibit 10.24 to the Company’s Annual Report on Form 10-K, filed on February 28, 2017). | ||||||
| 10.17 | Modification No. 1, effective January 27, 2017, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.22 to the Company's Annual Report on Form 10-K filed on February 23, 2018). | |||||||
| 10.18 | † | Modification No. 2, effective February 23, 2017, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.23 to the Company's Annual Report on Form 10-K filed on February 23, 2018). | ||||||
| 10.19 | Modification No. 3, effective March 22, 2017, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.24 to the Company's Annual Report on Form 10-K filed on February 23, 2018). | |||||||
| 10.20 | † | Modification No. 4, effective April 5, 2017, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.25 to the Company's Annual Report on Form 10-K filed on February 23, 2018). | ||||||
| 10.21 | † | Modification No. 5, effective September 8, 2017, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q filed on November 3, 2017). | ||||||
| 10.22 | † | Modification No. 6, effective September 21, 2017, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.27 the Company’s Annual Report on Form 10-K filed on February 23, 2018). | ||||||
| 10.23 | † | Modification No. 7, effective February 26, 2018, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q filed on May 4, 2018). | ||||||
| 10.24 | Modification No. 8, effective March 6, 2018, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q filed on May 4, 2018). | |||||||
| 10.25 | † | Modification No. 9, effective June 6, 2018, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q filed on August 3, 2018). | ||||||
| 10.26 | † | Modification No. 10, effective June 18, 2018, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q filed on August 3, 2018). | ||||||
| 10.27 | † | Modification No. 11, effective June 20, 2018, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q filed on August 3, 2018). | ||||||
| 10.28 | † | Modification No. 12, effective June 21, 2018, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q filed on August 3, 2018). | ||||||
| 10.29 | † | Modification No. 13, effective September 21, 2018 to the CDC BioThrax Procurement (incorporated by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q filed on November 2, 2018). | ||||||
| 10.30 | † | Modification No. 14, effective October 1, 2018, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.45 the Company’s Annual Report on Form 10-K filed on February 22, 2019). | ||||||
| 10.31 | † | Modification No. 15, effective December 7, 2018, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.46 the Company’s Annual Report on Form 10-K filed on February 22, 2019). | ||||||
| 10.32 | † | Modification No. 16, effective January 14, 2019, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.47 the Company’s Annual Report on Form 10-K filed on February 22, 2019). | ||||||
| 10.33 | †† | Modification No. 17, effective June 13, 2019, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q filed on August 2, 2019). | ||||||
| 10.34 | †† | Modification No. 18, effective September 11, 2019, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.39 the Company’s Annual Report on Form 10-K filed on February 25, 2020). | ||||||
| 10.35 | †† | Modification No. 19, effective January 6, 2020, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.40 the Company’s Annual Report on Form 10-K filed on February 25, 2020). | ||||||
| 10.36 | †† | Modification No. 20, effective January 7, 2020, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.41 the Company’s Annual Report on Form 10-K filed on February 25, 2020). | ||||||
| 10.37 | †† | Modification No. 21, effective January 7, 2020, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.45 the Company’s Annual Report on Form 10-K filed on February 19, 2021) | ||||||
| 10.38 | †† | Modification No. 22 to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.46 the Company’s Annual Report on Form 10-K filed on February 19, 2021) | ||||||
| 10.39 | †† | Modification No. 23, effective September 30, 2020, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.47 the Company’s Annual Report on Form 10-K filed on February 19, 2021) | ||||||
135
Exhibit Number | Exhibit Description | |||||||
| 10.40 | †† | (incorporated by reference to Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q filed on November 5, 2021). | ||||||
| 10.41 | †† | Modification No. 25, effective September 29, 2021, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.6 to the Company's Quarterly Report on Form 10-Q filed on November 5, 2021). | ||||||
| 10.42 | †† | Modification No. 26, effective November 1, 2021, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.48 the Company’s Annual Report on Form 10-K filed on February 25, 2022). | ||||||
| 10.43 | † | Modification No. 27, effective March 31, 2022, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q filed on April 29, 2022). | ||||||
| 10.44 | † | Modification No. 28, effective April 14, 2022, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q filed on August 2, 2022). | ||||||
| 10.45 | † | Modification No. 29, effective June 16, 2022, to the CDC BioThrax Procurement Contract (incorporated by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q filed on August 2, 2022). | ||||||
| 10.46 | Award/Contract, effective January 9, 2024 (BioThrax IDIQ Contract), from the United States Department of Defense to Emergent Biodefense Operations Lansing LLC (incorporated by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q filed on May 2, 2024). | |||||||
| 10.47 | Delivery Order 1, effective January 22, 2024 to the BioThrax IDIQ Contract (incorporated by reference to Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q filed on May 2, 2024). | |||||||
| 10.48 | Modification No. 1, effective February 13, 2024 to the BioThrax IDIQ Contract (incorporated by reference to Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q filed on May 2, 2024). | |||||||
| 10.49 | Modification No. 2, effective March 21, 2024 to the BioThrax IDIQ Contract (incorporated by reference to Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q filed on May 2, 2024). | |||||||
| 10.50 | Modification No. 3, effective December 13, 2024, to the BioThrax IDIQ Contract (incorporated by reference to Exhibit 10.2 to the Company's Current Report on Form 8-K filed January 8, 2025). | |||||||
| 10.51 | † | Award/Contract (the BARDA AV7909 Contract), effective September 30, 2016, from the BioMedical Advanced Research and Development Authority to Emergent Product Development Gaithersburg Inc. (incorporated by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q filed on November 9, 2016). | ||||||
| 10.52 | † | Modification No. 1, effective March 16, 2017, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q filed on November 5, 2021) | ||||||
| 10.53 | † | Modification No. 2, effective August 29, 2018, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q filed on November 5, 2021). | ||||||
| 10.54 | †† | Modification No. 3, effective July 30, 2019, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q filed on November 9, 2019). | ||||||
| 10.55 | †† | Modification No. 4, effective March 3, 2020, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q filed on May 1, 2020). | ||||||
| 10.56 | †† | Modification No. 5, effective April 10, 2020, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q filed on May 1, 2020). | ||||||
| 10.57 | †† | Modification No. 6, effective July 13, 2020, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q filed on November 6, 2020). | ||||||
| 10.58 | †† | Modification No. 7, effective December 2, 2020, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.1 to the Company's Quarterly Report on Form 10-Q filed on July 30, 2021). | ||||||
| 10.59 | †† | Modification No. 8, effective March 22, 2021, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.2 to the Company's Quarterly Report on Form 10-Q filed on July 30, 2021). | ||||||
| 10.60 | †† | Modification No. 9, effective April 21, 2021, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.3 to the Company's Quarterly Report on Form 10-Q filed on July 30, 2021). | ||||||
| 10.61 | †† | Modification No. 10, effective June 10, 2021 to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q filed on July 30, 2021). | ||||||
| 10.62 | †† | Modification No. 11, effective September 30, 2021, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q filed on November 5, 2021). | ||||||
| 10.63 | †† | Modification No. 12, effective December 2, 2021, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.61 to the Company's Annual Report on Form 10-K filed on February 25, 2022). | ||||||
| 10.64 | †† | Modification No. 13, effective March 30, 2023, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.4 to the Company's Quarterly Report on Form 10-Q filed on August 9, 2023). | ||||||
| 10.65 | †† | Modification No. 14, effective March 30, 2023, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q filed on August 9, 2023). | ||||||
136
Exhibit Number | Exhibit Description | |||||||
| 10.66 | † | Modification No. 15, effective October 6, 2023, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.80 to the Company's Annual Report on Form 10-K filed on March 8, 2024). | ||||||
| 10.67 | † | Modification No. 16, effective November 21, 2023, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.81 to the Company's Annual Report on Form 10-K filed on March 8, 2024). | ||||||
| 10.68 | †† | Modification No. 17, effective June 26, 2024, to the BARDA AV7909 Contract (incorporated by reference to Exhibit 10.2 to the Company's Current Report on Form 8-K, filed on July 2, 2024). | ||||||
| 10.69 | †† | Modification No. 18, effective December 12, 2024, to the BARDA AV909 Contract (incorporated by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K filed on January 8, 2025). | ||||||
| 10.70 | †† | |||||||
| 10.71 | †† | |||||||
| 10.72 | † | License Agreement, dated as of December 15, 2014, by and between Opiant Pharmaceuticals, Inc. (formerly known as Lightlake Therapeutics Inc.) and Adapt Pharma Operations Limited (incorporated by reference to Exhibit 10.76 to the Company's Annual Report on Form 10-K filed on March 4, 2025). | ||||||
| 10.73 | † | Amendment No. 1 to License Agreement, dated as of December 13, 2016, by and between Opiant Pharmaceuticals, Inc. and Adapt Pharma Operations Limited. (incorporated by reference to Exhibit 10.52 the Company’s Annual Report on Form 10-K filed on February 22, 2019). | ||||||
| 10.74 | † | Amendment No. 2 to License Agreement, dated December 15, 2014, by and between Opiant Pharmaceuticals, Inc. and Adapt Pharma Operations Limited, effective March 18, 2019 (incorporated by reference to Exhibit 10.1 the Company’s Quarterly Report on Form 10-Q filed on May 8, 2019). | ||||||
| 10.75 | †† | Award/Contract, effective August 30, 2019 (ACAM2000 Contract), from the Assistant Secretary, U.S. Department of Health and Human Services (ASPR/OPM) to Emergent Product Development Gaithersburg Inc. (incorporated by reference to Exhibit 10.48 the Company’s Annual Report on Form 10-K filed on February 25, 2020). | ||||||
| 10.76 | †† | Modification No. 1, effective, May 28, 2020 to the ACAM2000 Contract (incorporated by reference to Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q filed on July 31, 2020). | ||||||
| 10.77 | †† | Modification No. 2, effective, October 28, 2020 to the ACAM2000 Contract (incorporated by reference to Exhibit 10.60 the Company’s Annual Report on Form 10-K filed on February 19, 2021). | ||||||
| 10.78 | †† | Modification No. 3, effective, April 1, 2021 to the ACAM2000 Contract (incorporated by reference to Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q filed on July 30, 2021). | ||||||
| 10.79 | †† | Modification No. 4, effective, July 13, 2021 to the ACAM2000 Contract (incorporated by reference to Exhibit 10.69 to the Company's Annual Report on Form 10-K filed on February 25, 2022). | ||||||
| 10.80 | †† | Modification No. 5, effective, September 29, 2021 to the ACAM2000 Contract (incorporated by reference to Exhibit 10.70 to the Company's Annual Report on Form 10-K filed on February 25, 2022). | ||||||
| 10.81 | †† | Modification No. 6, effective, November 1, 2021 to the ACAM2000 Contract (incorporated by reference to Exhibit 10.71 to the Company's Annual Report on Form 10-K filed on February 25, 2022). | ||||||
| 10.82 | †† | Modification No. 7, effective October 6, 2022, to the ACAM2000 Contract (incorporated by reference to Exhibit 10.92 to the Company's Annual Report on Form 10-K filed on March 8, 2024). | ||||||
| 10.83 | † | Modification No. 8, effective November 21, 2022, to the ACAM2000 Contract (incorporated by reference to Exhibit 10.93 to the Company's Annual Report on Form 10-K filed on March 8, 2024). | ||||||
| 10.84 | † | Modification No. 9, effective May 24, 2023, to the ACAM2000 Contract (incorporated by reference to Exhibit 10.6 to the Company's Quarterly Report on Form 10-Q filed on August 9, 2023). | ||||||
| 10.85 | † | Modification No. 10, effective May 26, 2023, to the ACAM2000 Contract (incorporated by reference to Exhibit 10.7 to the Company's Quarterly Report on Form 10-Q filed on August 9, 2023). | ||||||
| 10.86 | †† | Modification No. 11, effective April 29, 2024, to the ACAM2000 Contract (incorporated by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K, filed on May 1, 2024). | ||||||
| 10.87 | †† | Modification No. 12, effective June 28, 2024, to the ACAM2000 Contract (incorporated by reference to Exhibit 10.1 to the Company's Current Report on Form 8-K, filed on July 2, 2024). | ||||||
| 10.88 | †† | Work Order to Manufacturing Services Agreement, dated June 10, 2020, between Emergent Manufacturing Operations Baltimore, LLC and AstraZeneca Pharmaceuticals LP (included as part of AZ MSA) (incorporated by reference to Exhibit 10.14 to the Company's Quarterly Report on Form 10-Q filed on November 6, 2020). | ||||||
| 10.89 | †† | Amendment No. 1, effective September 30, 2020, to AZ MSA (incorporated by reference to Exhibit 10.15 to the Company's Quarterly Report on Form 10-Q filed on November 6, 2020). | ||||||
| 10.90 | †† | |||||||
137
Exhibit Number | Exhibit Description | |||||||
| 10.91 | †† | |||||||
| 10.92 | †† | Amendment No. 2, effective October 30, 2020, to AZ MSA (incorporated by reference to Exhibit 10.5 to the Company's Quarterly Report on Form 10-Q filed on April 30, 2021). | ||||||
| 10.93 | †† | Amendment No. 3, effective November 25, 2020, to AZ MSA (incorporated by reference to Exhibit 10.6 to the Company's Quarterly Report on Form 10-Q filed on April 30, 2021). | ||||||
| 10.94 | †† | Amendment No. 4, effective January 21, 2021, to AZ MSA (incorporated by reference to Exhibit 10.7 to the Company's Quarterly Report on Form 10-Q filed on April 30, 2021). | ||||||
| 10.95 | †† | Change Order No. 1 to Work Order #5997-01, effective July 31, 2020, to AZ MSA (incorporated by reference to Exhibit 10.11 to the Company's Quarterly Report on Form 10-Q filed on July 30, 2021). | ||||||
| 10.96 | †† | Change Order No. 2 to Work Order #5997-01, effective August 04, 2020, to AZ MSA (incorporated by reference to Exhibit 10.12 to the Company's Quarterly Report on Form 10-Q filed on July 30, 2021). | ||||||
| 10.97 | †† | Change Order No. 4 to Work Order #5997-01, effective November 17, 2020, to AZ MSA (incorporated by reference to Exhibit 10.13 to the Company's Quarterly Report on Form 10-Q filed on July 30, 2021). | ||||||
| 10.98 | †† | Change Order No. 5 to Work Order #5997-01, effective September 16, 2020, to AZ MSA (incorporated by reference to Exhibit 10.14 to the Company's Quarterly Report on Form 10-Q filed on July 30, 2021). | ||||||
| 10.99 | †† | Change Order No. 6 to Work Order #5997-01, effective October 13, 2020, to AZ MSA (incorporated by reference to Exhibit 10.15 to the Company's Quarterly Report on Form 10-Q filed on July 30, 2021). | ||||||
| 10.100 | †† | Change Order No. 10 to Work Order #5997-01, effective March 10, 2021, to AZ MSA (incorporated by reference to Exhibit 10.16 to the Company's Quarterly Report on Form 10-Q filed on July 30, 2021). | ||||||
| 10.101 | †† | Change Order No. 13 to Work Order #5997-01, effective April 23, 2021, to AZ MSA (incorporated by reference to Exhibit 10.17 to the Company's Quarterly Report on Form 10-Q filed on July 30, 2021). | ||||||
| 19 | # | |||||||
| 21 | # | |||||||
| 23 | # | |||||||
| 31.1 | # | |||||||
| 31.2 | # | |||||||
| 32.1 | # | |||||||
| 32.2 | # | |||||||
| 97.1 | # | |||||||
| 101 | # | The following financial information related to the Company’s Annual Report on Form 10-K for the year ended December 31, 2025, formatted in iXBRL (Inline Extensible Business Reporting Language): (i) the Consolidated Balance Sheets, (ii) the Consolidated Statements of Operations, (iii) the Consolidated Statements of Comprehensive Income (Loss), (iv) the Consolidated Statements of Cash Flows, (v) the Consolidated Statement of Changes in Stockholders’ Equity; (vi) the related Notes to Consolidated Financial Statements; and (vii) the Cover Page. | ||||||
| 104 | # | Cover Page Interactive Data File, formatted in iXBRL and contained in Exhibit 101. | ||||||
| # | Filed herewith | |||||||
| † | Confidential treatment granted by the SEC as to certain portions. Confidential materials omitted and filed separately with the SEC. | |||||||
| †† | Certain confidential portions of this exhibit were omitted by means of marking such portions with asterisks because the identified confidential portions (i) are not material and (ii) are items the Company customarily and actually treats such information as private or confidential. | |||||||
| * | Management contract or compensatory plan or arrangement filed herewith in response to Item 15(a) of Form 10-K. | |||||||
138
ITEM 16. FORM 10-K SUMMARY
Not applicable.
139
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| EMERGENT BIOSOLUTIONS INC. | |||||
By: /s/RICHARD S. LINDAHL | |||||
| Richard S. Lindahl | |||||
Executive Vice President, Chief Financial Officer | |||||
| Date: February 26, 2026 | |||||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Signature | Title | Date | ||||||||||||
/s/Joseph C. Papa Joseph C. Papa | President, Chief Executive Officer and Director (Principal Executive Officer) | February 26, 2026 | ||||||||||||
/s/Richard S. Lindahl Richard S. Lindahl | Executive Vice President, Chief Financial Officer (Principal Financial and Accounting Officer) | February 26, 2026 | ||||||||||||
/s/Zsolt Harsanyi, Ph.D. Zsolt Harsanyi, Ph.D. | Director | February 26, 2026 | ||||||||||||
/s/Sujata Dayal Sujata Dayal | Director | February 26, 2026 | ||||||||||||
/s/Donald DeGolyer Donald DeGolyer | Director | February 26, 2026 | ||||||||||||
/s/Neal Fowler Neal Fowler | Director | February 26, 2026 | ||||||||||||
/s/Keith Katkin Keith Katkin | Director | February 26, 2026 | ||||||||||||
/s/Ronald B. Richard Ronald B. Richard | Director | February 26, 2026 | ||||||||||||
/s/Marvin White Marvin White | Director | February 26, 2026 | ||||||||||||
/s/Kathryn Zoon, Ph.D. Kathryn Zoon, Ph.D. | Director | February 26, 2026 | ||||||||||||
140

Effective Global Policy Insider Trading Policy 1 Global Policy : Insider Trading Policy Insider Trading Policy Purpose & Scope The purpose of this Policy is to: Provide general guidelines to all members of the Board of Directors (“Board Members”), officers, employees and designated consultants of Emergent BioSolutions Inc., subsidiaries and affiliates (collectively, “Emergent”) with respect to trading in Emergent securities, as well as the securities of publicly traded companies with whom Emergent has a business relationship. This Policy’s scope applies to: Board Members, officers, employees and designated consultants of Emergent, including employees and designated consultants of subsidiaries located outside of the United States (any of the foregoing sometimes referred to as “you” or “your”). Members of your immediate family (including domestic partners and adopted or step-children) and others who reside with you (but excluding unrelated tenants and household employees), as well as any family members who do not live in your household but whose transactions in Emergent securities are directed by you or are subject to your influence or control (such as family members who consult with you before they trade in Emergent securities) (collectively, “Family Members”). All corporations, limited liability companies, partnerships, trusts, investment funds and other entities controlled by you or any Family Member (the “Controlled Entities”) and transactions by these Controlled Entities should be treated for the purposes of this Policy and applicable securities laws as if they were for your own account. You should consult with Emergent’s Legal Department via email at Legal@ebsi.com if you are affiliated with an entity that you believe might be considered to be a Controlled Entity that may trade in Emergent’s securities. All of the individuals and entities that are covered by this Policy are sometimes referred to herein collectively as “Covered Persons”. Policy Owner: Jessica Perl Policy Sponsor: Jessica Perl Policy Contact: StockTransaction@ebsi.com Last updated: 29 Oct 2025

Effective Global Policy Insider Trading Policy 2 Introduction This Policy has been designed to prevent violations of insider trading laws. This Policy prohibits both illegal activities and trading activities that may not necessarily be illegal. Your strict adherence to this Policy will help safeguard the reputation of Emergent and its directors, officers and employees. Each Board Member, officer, employee and designated consultant of Emergent is responsible for the consequences of his or her actions. You are responsible for understanding and complying with this Policy. You are responsible for making sure that any transaction in securities covered by this Policy by any Family Member or Controlled Entity complies with this Policy. Any person who has a question about this Policy or its application to any proposed transaction may obtain additional guidance from Stock Plan Administration, who can be reached by e-mail at StockTransaction@ebsi.com. Policy Guiding Principles Principle 1: Know the law, how it is enforced and the penalities associated with illegal insider trading Securities Laws Federal securities laws, and certain applicable state securities laws, prohibit the purchase or sale of a company’s securities by anyone who is aware of material non-public information about that company. These laws also prohibit anyone who is aware of material non-public information from disclosing this information to others who may trade on the basis of such information. Companies and their controlling persons may also be subject to liability if they fail to take reasonable steps to prevent improper insider trading by company personnel. “Securities” include common stock, debt securities, preferred stock and derivative securities such as options to purchase common stock (including put and call options), warrants, swaps, futures and forward contracts. Transactions in securities include purchases, sales, pledges, hedges and loans of securities, as well as other direct or indirect transfers of securities. Certain of these transactions are addressed in more detail herein and may not be permitted under this Policy even if you are not aware of material non-public information about the issuer of the securities in question.

Effective Global Policy Insider Trading Policy 3 Enforcement and Penalties Violations of applicable insider trading laws can result in severe civil and criminal penalties under U.S. federal and state law. For example, under U.S. federal securities laws, insider trading violations may subject individuals to imprisonment for up to 20 years, criminal fines of up to $5 million and civil fines of up to three times the profit gained or loss avoided. In addition, Emergent could be subject to civil and criminal fines as a result of the insider trading violations of others. Failure to comply with this Policy may also subject you to disciplinary actions imposed by Emergent, up to and including disgorgement of proceeds and termination for cause, whether or not your failure to comply with this Policy results in a violation of law. It is important that you understand the breadth of activities that constitute illegal insider trading and the consequences, which can be severe. Cases have been successfully prosecuted involving trading through foreign accounts, trading by family members and friends, and trading involving only a small number of shares. Both the U.S. Securities and Exchange Commission (the “SEC”) and the Financial Industry Regulatory Authority, Inc. (“FINRA”) investigate and are very effective at detecting insider trading. Both the SEC and the U.S. Department of Justice pursue insider trading violations vigorously. Principle 2: Be aware of and alert to what is considered material non-public information Material Information Information about a company or its securities is “material” if a reasonable investor would likely consider it important in making a decision to buy, hold or sell securities. Any information that could reasonably be expected to affect the market price of a security is material. The information may be positive or negative. Financial information of a company is frequently material, even if it covers only part of a fiscal period or less than all of the company’s operations, because either of these might convey enough information about the company’s consolidated results to be considered material information. Common examples of information that may be material include: Information about quarterly or annual revenues, liquidity, or information about material impacts to quarterly or annual revenues of specific products; Projections of future earnings or losses, or other financial guidance or changes thereto, including an anticipated decision to suspend financial guidance; A significant pending procurement contract for a product or a significant pending development contract for a product candidate that the company reasonably anticipates will generate substantial future revenues; An imminent significant licensing transaction, collaboration, merger, acquisition or disposition of assets (e.g. where the term sheet related to a proposed transaction has been signed); A significant company restructuring, board or management change; Significant regulatory developments involving the U.S. Food and Drug Administration or foreign regulatory agencies, and other regulatory developments regarding a product, anticipated regulatory approvals material to the company or information regarding a potential product recall;

Effective Global Policy Insider Trading Policy 4 Advanced knowledge of changes to key officials at government agencies with which the company does business; Significant planned bank borrowings or other financing transactions out of the ordinary course; A change in the company’s dividend policy, the declaration of a stock split, or a planned offering of securities; The establishment of a stock repurchase program; Pending or threatened significant litigation, or the resolution of such litigation; A notice of issuance of material patents related to the company’s products or pipeline; A significant cybersecurity incident; A change in auditors or notification that the auditors’ reports may no longer be relied upon; Significant changes in the company’s overall strategy or marketing plan for its products; Significant changes in the company’s manufacturing capabilities; and The gain or loss of a significant contract, customer, or supplier. The above list does not include all of the information that could be deemed to be material. It is not possible to define all categories of material information, and you should recognize that the public, the media, regulatory agencies and the courts will use hindsight in judging what is material. Moreover, whether information is material must be considered in the context of all facts and circumstances at a given time. Therefore, it is important to err on the safe side and assume information is material if there is any doubt. Non-Public Information Information is “non-public” if it has not been widely disseminated to the public. Information may still be non-public even though it is widely known within Emergent. Release of information to the media does not immediately mean the information has become publicly available. Information is considered to be widely disseminated to the public only when it has been released broadly to the marketplace (such as by a press release carried over a major news service or an SEC filing) and the investing public has had time to absorb and evaluate it. The fact that non-public information is reflected in rumors in the marketplace does not mean that the information has been widely disseminated. It is important to note that even after information becomes public, many aspects relating to a matter may remain non-public. Ordinarily, information about Emergent should not be considered public until at least one full trading day has passed following its formal release to the market.

Effective Global Policy Insider Trading Policy 5 Applying the above guidance in a practical example: If Emergent discloses material non-public information before trading begins on a Tuesday and you have prior knowledge of such material non-public information, the first time you can buy or sell Emergent securities is the opening of the market on Wednesday (assuming you are not aware of other material non-public information at that time). If, however, Emergent discloses the material non-public information after trading begins that Tuesday, the first time you can buy or sell Emergent securities is the opening of the market on Thursday. Keep in mind that, even if material non-public information has been widely disseminated to the public and there has been sufficient time for the investing public to absorb and evaluate such information, your ability to trade in Emergent securities may be restricted by a blackout period, as discussed below. Principle 3: Covered Persons are prohibited from trading in Emergent securities while aware of material non-public information about Emergent Subject to certain limited exceptions, as described below, Covered Persons are prohibited from transacting in Emergent securities, or having others engage in such transactions on your behalf, while aware of material non-public information about Emergent or recommending to another person that they do so. It makes no difference whether or not you relied upon or used material non-public information in deciding to trade – if you are aware of material non-public information about Emergent, the prohibition applies, even if the proposed trade would not take place during a blackout period or if you previously received pre-clearance for the trade. You should avoid even the appearance of an improper transaction to preserve Emergent’s reputation for adhering to the highest ethical standards of conduct. The following are examples of common transactions and information about whether the trading prohibition would apply. Stock Option Exercises The trading prohibition does not apply to the exercise of stock options issued under Emergent benefit plans if the exercise price is paid in cash or through Emergent withholding a portion of the shares underlying the options. Similarly, Emergent may withhold underlying shares to satisfy tax withholding requirements upon the exercise of stock options. This prohibition does apply, however, to sales of the underlying stock and broker-assisted cashless exercises of options, as well as to any other market sales of Emergent securities for the purpose of generating the cash needed to cover the costs of exercise or tax withholding obligations. Vesting of Restricted Stock Units or Settlement of Performance Stock Units The trading prohibition does not apply to the automatic deduction of shares by Emergent from your restricted stock unit or performance stock unit account to satisfy the minimum statutory tax withholding liability upon the vesting of restricted stock units or settlement of performance stock units. The prohibition does apply, however, to any open market sale of vested shares, including to satisfy tax liabilities.

Effective Global Policy Insider Trading Policy 6 Rule 10b5-1 Plans and Non-Rule 10b5-1 Trading Arrangements The trading prohibition does not apply to trades made pursuant to a valid Rule 10b5-1 Plan or Non-Rule 10b5-1 Trading Arrangement approved by Emergent as described below. Employee Stock Purchase Plan The trading prohibition does not apply to automatic purchases of Emergent securities in the employee stock purchase plan resulting from your periodic contribution of money to the plan during open enrollment or purchases under the plan that were made pursuant to instructions that you provided when you were not aware of material non- public information. This prohibition does apply, however, to (i) any election to participate in the plan for an enrollment period, (ii) increases or decreases in your amount of periodic contribution to the plan, and (iii) your sales of common stock purchased pursuant to the plan. 401(k) and Other Individual Account Plans The trading prohibition applies to the acquisition or disposition of Emergent securities in individual retirement account plans in the same manner as it applies to any other purchase or sale of Emergent’s securities. Emergent’s 401(k) plan does not permit the holding of Emergent’s securities in an individual’s 401(k) or other Emergent- sponsored retirement account. Transactions Not Involving a Purchase or Sale Bona fide gifts of Emergent securities are not transactions subject to this Policy. You should be aware, however, that gifts are not allowed to circumvent the insider trading laws, so it is important to consider whether you possess material non-public information before making or receiving a bona fide gift, particularly if it is likely that the recipient of the gift is likely to sell the securities in the near term. In addition, if the person making the gift is subject to the pre-clearance procedures specified below, then such procedures must be followed in connection with the gift. Depending on the circumstances, recipients of gifts may be subject to restrictions on subsequent sales of securities. Principle 4: Covered Persons cannot trade in securities of other companies while aware of material non-public information about those companies Emergent may engage in business relationships or transactions with companies whose securities are publicly traded. These transactions may include, among other things, mergers, acquisitions, divestitures, manufacturing, supply and distribution arrangements or the renewal or termination of significant contracts or other arrangements. In addition, Emergent has relationships with various customers, distributors, manufacturers, suppliers and other firms, and there may be other companies with which trading in Emergent’s securities is highly correlated. Information learned in connection with these transactions or relationships may constitute material non-public information about the other company. Covered Persons are prohibited from trading in the securities of these companies while aware of material non-public information about the companies and from providing such information to any other person or recommending that they trade in such companies’ securities.

Effective Global Policy Insider Trading Policy 7 Principle 5: ‘Tipping’ of material non-public information is illegal; refrain from recommending transactions in securities Covered Persons may not pass material non-public information about Emergent or any other company on to any other person if such person may engage in a purchase or sale on the basis of that information, regardless of whether you profit or intend to profit by the tipping, disclosure, or use. This practice, known as “tipping”, also violates the securities laws and can result in the same civil and criminal penalties that apply to insider trading, even if you did not trade or gain any benefit from another person's trading. Your duty to maintain as confidential all material non- public information about Emergent or its business partners is addressed in further detail below. Covered Persons also may not recommend or suggest to others that they trade such company’s securities or provide advice of any kind about such company or its securities. Principle 6: Material non-public information must be kept confidential Material non-public information about Emergent, its business partners or companies with which trading in Emergent securities is highly correlated, should be maintained in strict confidence and should be discussed, even within Emergent, only with persons who have a need to know in order to perform their legitimate business duties. Covered Persons should exercise the utmost care and circumspection in dealing with information that may be material non- public information. In addition, Covered Persons must maintain in strict confidence all proprietary information of Emergent, even if it does not constitute material non-public information. Conversations in public places, such as hallways, elevators, restaurants and airplanes, involving information of a sensitive or confidential nature should be avoided. Written information should be appropriately safeguarded and should not be left where it may be seen by persons not entitled to the information. The unauthorized disclosure of information could result in serious consequences to Emergent, whether or not the disclosure is made for the purpose of facilitating improper trading in securities. Principle 7: Be aware of all the restrictions placed on Covered Persons when trading in Emergent securities Frequent trading of Emergent securities is strongly discouraged Frequent trading of Emergent securities can create an appearance of wrongdoing even if the decision to trade was based solely on public information. Covered Persons are strongly discouraged from trading in Emergent securities for short-term trading profits. Daily or frequent trading, which can be time-consuming and distracting, is strongly discouraged. Emergent reserves the right to require you to provide brokerage account statements to assure compliance with this and other provisions of this Policy.

Effective Global Policy Insider Trading Policy 8 No short sales of Emergent securities Covered Persons may not engage in short sales of Emergent securities (sales of securities that are not then owned), including “sales against the box” (short sales not exceeding the number of shares already owned, but prior to the closing out of an existing “long” position). Generally, short sales are transactions whereby a person will benefit from a decline in the price of the securities, and Emergent believes it is inappropriate for Covered Persons to engage in these transactions with respect to Emergent securities. No trading in derivatives of Emergent Covered Persons may not trade in derivatives of an Emergent security, such as exchange-traded put or call options and forward transactions. No hedging transactions Certain forms of hedging or monetization transactions may offset a decrease in the value of Emergent securities you hold, enabling you to continue to own Emergent securities without the full risks and rewards of ownership. Emergent believes that such transactions separate the holder's interests from those of other stockholders. Therefore, Covered Persons and any person acting on a Covered Person’s behalf are prohibited from purchasing any financial instruments (such as prepaid variable forward contracts, collars, equity swaps and put options) or otherwise engaging in any transactions that hedge or offset, or are designed to hedge or offset, any decrease in the market value of Emergent securities. No margin accounts or pledges without prior approval Securities held in a margin account or pledged as collateral for a loan may be sold without your consent by the broker if you fail to meet a margin call or by the lender in foreclosure if you default on the loan. Because a margin or foreclosure sale may occur at a time when a Covered Person is aware of material non-public information or otherwise are not permitted to trade in Emergent securities, Covered Persons are prohibited from holding Emergent securities in a margin account or pledging Emergent securities as collateral for a loan, unless approved in advance by the General Counsel in writing. Requests for approval must be submitted at least two weeks in advance of the intended pledge date. Pledges during blackout periods are prohibited under this Policy. Limited use of standing orders A standing order placed with a broker to sell or purchase stock at a specified price should be used for a maximum of five business days. If you are a Restricted Person (as defined below) subject to the trading pre-clearance requirement and determine that you must use a standing order or limit order on Emergent securities, the order should be limited to no longer than the approved trading authorization period (i.e., a pre-clearance will last until the earlier of the five-day trading authorization or the beginning of a Quarterly Blackout Period or Special Blackout Period, as each are described below.

Effective Global Policy Insider Trading Policy 9 A standing order placed with a broker to sell or purchase stock at a specified price leaves you with no control over the timing of the transaction. A standing order transaction executed by the broker when you are aware of material non-public information may result in unlawful insider trading. Accordingly, a Covered Person may not place a standing order transaction with a broker with respect to an Emergent security while in possession of material non- public information concerning Emergent. However, a standing order incorporated into a Rule 10b5-1 Plan or Non- Rule 10b5-1 Trading Arrangement approved by Emergent is permitted. No trading on rumors Rumors within Emergent concerning matters which, if true, would be material non-public information are deemed to constitute material non-public information for purposes of this Policy. Accordingly, Covered Persons should not trade on the basis of these rumors. Post-termination transactions may be prohibited The portions of this Policy prohibiting trading while in possession of material non-public information and the use or disclosure of that information continue to apply to transactions in Emergent securities and those of its business partners and other companies, even after a Covered Person’s termination of employment or association with Emergent. If you are aware of material non-public information about Emergent or any such other company when your employment or other business relationship with Emergent ends, you may not trade in Emergent securities or those of such other companies or disclose the material non-public information to anyone else until that information is broadly disseminated to the public or ceases to be material. Principle 8: Restricted Persons are subject to Quarterly Blackout Periods; Designated Persons are Restricted Persons who are also subject to Quarterly Blackout Periods as well as Pre-Clearance requirements; Special Blackout Periods can apply to any Covered Person “Restricted Persons” are those who, in the view of Emergent, are at an enhanced risk of possessing material non- public information and who therefore must exercise greater diligence to comply with insider trading prohibitions. This group includes all Board Members, officers and certain finance, legal, investor relations, corporate communications and management employees, as well as any other employees in a role that makes it likely they will have regular access to material non-public financial information. The list of Restricted Persons is updated on a quarterly basis by Stock Plan Administration in consultation with the General Counsel’s office, Finance and Human Resources. You will be notified by Stock Plan Administration if you are considered a Restricted Person under this Policy and subject to Quarterly Blackout Periods and, in certain cases, pre-clearance requirements, as explained below.

Effective Global Policy Insider Trading Policy 10 “Designated Persons” are certain Restricted Persons who are designated by the General Counsel, or his or her designee, in consultation with the Chief Financial Officer, as applicable, from time to time, and who must obtain pre- clearance by Stock Plan Administration before engaging in any transaction involving Emergent securities, including, but not limited to, purchases, sales, and gifts. Board Members, Senior Vice President-level employees and above and certain other designated employees of Emergent are “Designated Persons,” as well as their Family Members and Controlled Entities, and may not engage in any transaction in Emergent securities without first obtaining pre-clearance of the transaction from Stock Plan Administration. The pre-clearance process is described below. You will be notified by Stock Plan Administration if you are considered a Designated Person under this Policy. All Designated Persons shall also be deemed to be and subject to the additional restrictions placed on Restricted Persons. Quarterly Blackout Periods With respect to each quarterly earnings announcement, a “Quarterly Blackout Period” is in effect starting on the 15th day of the third month of the applicable Emergent fiscal quarter and ending when one full trading day has passed following the public announcement of Emergent’s quarterly financial results. Emergent has selected this period because it is the time when there is likely to be material non-public information about Emergent that may be available to Restricted Persons. If you are a Restricted Person, you may not trade during a Quarterly Blackout Period, regardless of whether you are then actually aware of material non-public information about Emergent, except pursuant to a valid pre-existing Rule 10b5-1 Plan or Non-Rule 10b5-1 Trading Arrangement approved by Emergent. See additional details in Part C below. Although you are always responsible for monitoring for yourself whether you possess material non-public information, from time to time Emergent may decide to impose a special trading blackout on those who are aware of particular information that Emergent determines may be considered material non-public information (a “Special Blackout Period”). This kind of trading blackout may be imposed in connection with a significant event or development. If you are subject to a Special Blackout Period, you may not trade in any Emergent securities, except pursuant to a Rule 10b5-1 Plan or Non-Rule 10b5-1 Trading Arrangement previously approved by Emergent, until notified that the blackout has ended. Notwithstanding the above, a Quarterly Blackout Period does not prohibit trading in Emergent securities pursuant to a valid pre-existing Rule 10b5-1 Plan or Non-Rule 10b5-1 Trading Arrangement approved by Emergent as described below. Special Blackout Periods The General Counsel, in consultation with the Chief Financial Officer, as applicable, will determine whether an event-specific blackout period (a “Special Blackout Period”) should be imposed and to whom it shall apply. The existence of a Special Blackout Period will not be generally announced. If you are covered by the Special Blackout Period, you will be notified by Stock Plan Administration. Any person made aware of a Special Blackout Period should not disclose the existence of the blackout to anyone else.

Effective Global Policy Insider Trading Policy 11 No person subject to a Special Blackout Period may trade in Emergent securities during the pendency of the blackout, regardless of whether they are then actually aware of material non-public information about Emergent. Persons covered by a Special Blackout Period must wait until at least one full trading day has passed after the public announcement of the event which triggered the Special Blackout Period. You will be notified by Stock Plan Administration once the Special Blackout Period has been lifted. Trading pre-clearance requirement for Designated Persons A Designated Person’s request for pre-clearance should be submitted to Stock Plan Administration at StockTransaction@ebsi.com at least two business days in advance of the proposed transaction. Stock Plan Administration is under no obligation to approve a transaction submitted for pre-clearance, and may determine not to permit the transaction. If a Designated Person seeks pre-clearance and permission to engage in the transaction is denied, then he or she should refrain from initiating any transaction in Emergent securities, and should not inform any other person of the restriction. To request pre-clearance, the requestor should complete the “Trading Pre-Clearance Form,” a form of which is attached hereto and which can be found on Emergent’s Intranet site, and submit the form to StockTransaction@ebsi.com. If a request for pre-clearance is approved, you will have five business days to effect the transaction (or, if earlier, until commencement of a Quarterly Blackout Period or Special Blackout Period). If the transaction is not completed within such time, pre-clearance must again be requested. Under no circumstances may a person trade while aware of material non-public information about Emergent, even if the transaction was pre- cleared. Thus, if you become aware of material non-public information after receiving pre-clearance, but before the trade has been executed, you must not effect the pre-cleared transaction. Emergent’s approval of any particular transaction under this pre-clearance procedure does not insulate any person from liability under the securities laws. Under the law, the ultimate responsibility for determining whether an individual is aware of material non- public information about Emergent rests with that person in all cases. For a summary of key dates and timing considerations described above, refer to the Key Dates and Timing Considerations Chart. Principle 9: Only pre-approved Rule 10b5-1 Plans and Non-Rule 10b5-1 Trading Arrangements allow for transactions to occur during Blackout Periods Transactions effected pursuant to a pre-approved Rule 10b5-1 Plan or Non-Rule 10b5-1 Trading Arrangement will not be subject to Emergent’s blackout periods or pre-clearance procedures as described above. These pre-planned trading programs are available only to Board Members, officers and such other Emergent employees as may be designated from time to time by the Chief Financial Officer or the General Counsel, as applicable.

Effective Global Policy Insider Trading Policy 12 It is recommended that you allow at least five business days for approval of a Rule 10b5-1 Plan from the General Counsel’s office. One of the factors that may be considered in determining whether to approve a Rule 10b5-1 Plan or Non-Rule 10b5-1 Trading Arrangement is compliance with Emergent’s applicable minimum stock ownership guidelines applicable to Board Members and executive officers, if applicable to you. For more information about how to establish a Rule 10b5-1 Plan or Non-Rule 10b5-1 Trading Arrangement, please contact Stock Plan Administration by e-mail at StockTransaction@ebsi.com. Rule 10b5-1 Plans Rule 10b5-1(c) of the Securities Exchange Act of 1934 permits corporate insiders to establish written trading plans (commonly referred to as “Rule 10b5-1 Plans”) that can be useful in enabling insiders to trade in their company’s securities with significantly reduced risk of insider trading liability. A valid Rule 10b5-1 Plan operates as an affirmative defense against a claim of insider trading for trades subsequently executed as specified by the plan, even if the insider is in possession of material non-public information at the time the trade is executed. A valid Rule 10b5-1 Plan must: consist of a plan, arrangement or instruction that satisfies the requirements of Rule 10b5-1; be documented in writing; be established in good faith and not as part of a plan or scheme to evade the prohibitions of Rule 10b5-1; if the insider is an officer or Board Member, include a certification that at the time of the adoption of the Rule 10b5-1 Plan, such person (1) is not aware of any material non-public information about Emergent or its securities and (2) is adopting the Rule 10b5-1 Plan in good faith and not as part of a plan or scheme to evade the prohibitions of Rule 10b5-1; prohibit the first trade under the Rule 10b5-1 Plan from occurring for (“cooling off” periods): o If the insider is an officer subject to Section 16 of the Securities Exchange Act of 1934, as amended (the “Section 16 Officer”) or Board Member, the longer of (1) 90 days following the adoption of the Rule 10b5- 1 Plan and (2) two business days following the disclosure in certain periodic reports of Emergent’s financial results for the fiscal quarter in which the plan was adopted (but not to exceed 120 days following adoption of the Rule 10b5-1 Plan); and o If the insider is not a Section 16 Officer or Board Member, at least 30 days after the adoption of the Rule 10b5-1 Plan; be established at a time that does not fall within a blackout period and when the insider does not possess any material non-public information; and be pre-approved in writing by the General Counsel’s office. In addition, an officer or Board Member adopting a new Rule 10b5-1 Plan may not have any other outstanding Rule 10b5-1 Plans, and may not enter into any additional Rule 10b5-1 Plans, unless otherwise approved by the General Counsel’s office, and only in transactions where such person acquires (or sells) securities through participation in employee stock ownership plans or dividend reinvestment plans, which are not executed by the person on the open market.

Effective Global Policy Insider Trading Policy 13 Note that any modification to the amount, pricing, or timing of purchases or sales of securities under a Rule 10b5-1 Plan will constitute the termination of the Rule 10b5-1 Plan and adoption of a new plan, which means that any such modification will trigger the need for the new trading plan to satisfy all of the requirements of Rule 10b5-1, including a new “cooling off” period, before trading can begin again. Non-Rule 10b5-1 Trading Arrangements The SEC has established criteria for trading arrangements that do not satisfy all of the criteria of Rule 10b5-1 but are still intended to operate as a defense to a claim of insider trading (“Non-Rule 10b5-1 Trading Arrangements”). These criteria are set forth in Item 408 of the SEC’s Regulation S-K. Non-Rule 10b5-1 Trading Arrangements are subject to similar procedural requirements as Rule 10b5-1 Plans but do not involve “cooling off” periods and are not subject to restrictions on overlapping plans. Disclosure Related to 10b5-1 Trading Arrangements The Company shall require disclosure in SEC Forms 4 or 5 whether a transaction was made pursuant to a 10b5-1 Trading Arrangement. The adoption, amendment, or termination of a 10b5-1 Trading Arrangement shall be publicly disclosed in the Company’s quarterly filings. Principle 10: Participation in communications via social media, text chats, group messenger apps, electronic bulletin boards, chat rooms, blogs or websites must be consistent with this Policy Any written or verbal statement that would be prohibited under the law or under this Policy is equally prohibited if made on social media, in a text chat, group messenger app, electronic bulletin board, chat room, blog, or website, including the disclosure of material non-public information about Emergent or any other companies that you come into possession of as an employee, officer, Board Member or consultant of Emergent. Principle 11: Public disclosures should be made only by specified persons No individuals other than specifically authorized personnel should release material information to the public or respond to inquiries from the media, analysts, investors, or others outside of Emergent. You should refer any such inquires to both the global communications leader via email at mediarelations@ebsi.com or the Chief Financial Officer or head of investor relations via email at investorrelations@ebsi.com, as applicable. Roles and Responsibilities The General Counsel is responsible for appointing individuals to administer this Policy and its related pre-clearance procedures and a committee of representatives from each of Legal, Finance, Compliance and Human Resources (the “Resolution Team”) who shall be responsible for determining the consequences of any violations of this Policy.

Effective Global Policy Insider Trading Policy 14 All determinations and interpretations made by the Resolution Team shall be subject to the final review of the General Counsel, in his or her sole discretion. Exceptions In certain limited circumstances, a transaction otherwise prohibited by this Policy may be permitted if, prior to the transaction, the General Counsel determines that the transaction is not inconsistent with the purposes of this Policy. The existence of a personal financial emergency does not excuse any Covered Person from compliance with this Policy and will not be the basis for an exception to the Policy for a transaction that is inconsistent with the purposes of the Policy. An exception for a transaction otherwise prohibited by this Policy that is approved by the General Counsel shall only be approved in writing. References and Supporting Documents Emergent Code of Conduct & Business Ethics Policy External Communications Policy Exhibits Exhibit A: Trading Pre-Clearance Form Exhibit B: Key Dates and Timing Considerations Chart

Effective Global Policy Insider Trading Policy 15 Exhibit A: Trading Pre-Clearance Form EMERGENT BIOSOLUTIONS INC. TRADING PRE-CLEARANCE FORM DESCRIPTION OF PROPOSED TRANSACTION Name: Today's Date: Title: Department: Proposed Number of Shares: Type of Transaction: (Option Exercise, Purchase, Sale or Gift (describe)) CERTIFICATION: By my signature below, I certify to Emergent BioSolutions Inc. (the “Company”) that (1) I have read and am familiar with the Company’s Insider Trading Policy; (2) I am aware that it is unlawful to trade in the securities of the Company, or to enter into or modify a Rule 10b5-1 trading plan (“Trading Plan”), while in possession of material non-public information concerning the Company; (3) I am aware that civil and criminal penalties as well as damages could be imposed if I trade or enter into or modify a Trading Plan while in possession of material non- public information concerning the Company; (4) I am not in possession of or aware of any material information regarding the Company that has not been publicly disclosed by the Company in a widely disseminated press release or in a public filing with the SEC; and (5) In connection with evaluating my certification that I am not in possession of or aware of any material information regarding the Company that has not been publicly disclosed by the Company, I have considered whether I am aware of any Material Incident, as defined below, that has not been publicly disclosed, and certify that I am not in possession or aware of such information. “Material Incident is defined as a: (i) significant development in the Company’s relationships with key regulators, including, without limitation, any inspection with findings requiring changes, improvements, or reforms, inspection observations reported on a FDA Form 483, or material violations issued by the FDA or any other governmental or regulatory body; (ii) whistleblower or ethics complaint and the results of any investigations into such complaints that concern the facility, its operations, or a material contract the facility is responsible for; (iii) material and notable production issues, including, but not limited to: significant production delays, contamination issues, quality control, regulatory compliance, destruction of large quantities of products,

Effective Global Policy Insider Trading Policy 16 major personnel issues; or (iv) Contract disputes and significant developments in the Company’s relationships with key contractual partners, or production of their own vaccines or medications. Signature: Dated: FOR INTERNAL USE Approved? □ Yes □ No Date of Expiration of Clearance

Effective Global Policy Insider Trading Policy 17 Exhibit B: Key Dates and Timing Considerations Chart The chart below summarizes key dates and timing considerations from the Insider Trading Policy. Event Date of Occurrence Date for submission of pre-clearance request (Designated Persons only) Two business days in advance of a proposed transaction. Length of pre-clearance (Designated Persons only) Five business days to effect a transaction (or, if earlier, until commencement of a Quarterly Blackout Period or Special Blackout Period). Commencement of Quarterly Blackout Period (Restricted Persons only) 15th day of the third month of the applicable Emergent fiscal quarter. End of Quarterly Blackout Period (Restricted Persons only) One full trading day following the public announcement of Emergent’s quarterly financial results. Commencement of Special Blackout Period (notified persons only) As notified by Stock Plan Administration. End of Special Blackout Period (notified persons only) One full trading day after the public announcement of the event that triggered the Special Blackout Period, as notified by Stock Plan Administration. Date material non-public information is deemed to be public One full trading day following wide dissemination to the market. Use of a limit order Maximum of five business days (or, if earlier, until commencement of a Quarterly Blackout Period or Special Blackout Period).
List of Subsidiaries
(as of December 31, 2025)
Name of Subsidiary | Jurisdiction of Incorporation or Organization | ||||
Domestic | |||||
400 Professional LLC | Delaware | ||||
Cangene bioPharma LLC | Maryland | ||||
Emergent Commercial Operations Frederick Inc. | Maryland | ||||
Emergent Biodefense Operations Lansing LLC | Delaware | ||||
Emergent Devices Inc. | Delaware | ||||
Emergent Europe Inc. | Delaware | ||||
Emergent International Inc. | Delaware | ||||
Emergent Manufacturing Operations Baltimore LLC | Delaware | ||||
Emergent Product Development Gaithersburg Inc. | Delaware | ||||
Emergent Travel Health Inc. | Delaware | ||||
International | |||||
Emergent Acquisition Unlimited Company | Ireland | ||||
Emergent BioSolutions Canada Inc. | Ontario | ||||
Emergent BioSolutions Ireland Limited | Ireland | ||||
*Emergent Netherlands B.V. | Netherlands | ||||
Emergent Operations Ireland Limited | Ireland | ||||
Emergent Sales and Marketing Australia Pty Ltd. | Australia | ||||
Emergent Sales and Marketing Germany GmbH | Germany | ||||
Emergent Sales and Marketing Singapore Pte. Ltd. | Singapore | ||||
Emergent BioSolutions UK Ltd. | England and Wales | ||||
*The Company chose not to proceed with the dissolution of this entity indicated in FY25.
Exhibit 23
Consent of Independent Registered Public Accounting Firm
We consent to the incorporation by reference in the following Registration Statements:
(1)Registration Statement (Form S-3 No. 333-289359) of Emergent Biosolutions Inc.
(2)Registration Statement (Form S-8 No. 333-196232) pertaining to the Third Amended and Restated Emergent BioSolutions Inc. 2006 Stock Incentive Plan,
(3)Registration Statement (Form S-8 No. 333-216294) pertaining to the Fourth Amended and Restated Emergent BioSolutions Inc. 2006 Stock Incentive Plan,
(4)Registration Statement (Form S-8 No. 333-225283) pertaining to the Emergent BioSolutions Inc. Stock Incentive Plan,
(5)Registration Statement (Form S-8 No. 333-256798) pertaining to the Emergent BioSolutions Inc. Amended and Restated Stock Incentive Plan,
(6)Registration Statement (Form S-8 No. 333-272433) pertaining to the Emergent BioSolutions Inc. Amended and Restated Stock Incentive Plan and the Emergent BioSolutions Inc. Amended Employee Stock Purchase Plan,
(7)Registration Statement (Form S-8 POS No. 333-275990) pertaining to the Emergent BioSolutions Inc. Inducement Plan,
(8)Registration Statement (Form S-8 No. 333-281292) pertaining to the Emergent BioSolutions Inc. Amended and Restated Stock Incentive Plan,
(9)Registration Statement (Post Effective Amendments No. 1 and No. 2 to Form S-1 No. 333-283150) of Emergent BioSolutions Inc.
of our reports dated February 26, 2026, with respect to the consolidated financial statements and schedule of Emergent BioSolutions Inc. and subsidiaries and the effectiveness of internal control over financial reporting of Emergent BioSolutions Inc. and subsidiaries included in this Annual Report (Form 10-K) of Emergent BioSolutions Inc. and subsidiaries for the year ended December 31, 2025.
/s/ Ernst & Young LLP
Tysons, Virginia
February 26, 2026
EXHIBIT 31.1
CERTIFICATION
I, Joseph C. Papa, certify that:
(1) I have reviewed this Annual Report on Form 10-K of Emergent BioSolutions Inc.;
(2) Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
(3) Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
(4) The registrant's other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the registrant's disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the registrant's internal control over financial reporting that occurred during the registrant's most recent fiscal quarter (the registrant's fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant's internal control over financial reporting; and
(5) The registrant's other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant's auditors and the audit committee of the registrant's board of directors (or persons performing the equivalent functions):
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant's ability to record, process, summarize and report financial information, and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant's internal control over financial reporting.
Date: February 26, 2026
/s/Joseph C. Papa
Joseph C. Papa
President and Chief Executive Officer
EXHIBIT 31.2
CERTIFICATION
I, Richard S. Lindahl, certify that:
(1) I have reviewed this Annual Report on Form 10-K of Emergent BioSolutions Inc.;
(2) Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
(3) Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
(4) The registrant's other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the registrant's disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the registrant's internal control over financial reporting that occurred during the registrant's most recent fiscal quarter (the registrant's fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant's internal control over financial reporting; and
(5) The registrant's other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant's auditors and the audit committee of the registrant's board of directors (or persons performing the equivalent functions):
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant's ability to record, process, summarize and report financial information, and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant's internal control over financial reporting.
Date: February 26, 2026
/s/RICHARD S. LINDAHL
Richard S. Lindahl
Chief Financial Officer
EXHIBIT 32.1
CERTIFICATION PURSUANT TO 18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report on Form 10-K of Emergent BioSolutions Inc. (the "Company") for the period ended December 31, 2025 as filed with the Securities and Exchange Commission on the date hereof (the "Report"), the undersigned, Joseph C. Papa, President and Chief Executive Officer of the Company, hereby certifies, pursuant to 18 U.S.C. Section 1350, that:
(1) The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
(2) The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
Date: February 26, 2026
/s/Joseph C. Papa
Joseph C. Papa
President and Chief Executive Officer
EXHIBIT 32.2
CERTIFICATION PURSUANT TO 18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Annual Report on Form 10-K of Emergent BioSolutions Inc. (the "Company") for the period ended December 31, 2025 as filed with the Securities and Exchange Commission on the date hereof (the "Report"), the undersigned, Richard S. Lindahl, Chief Financial Officer of the Company, hereby certifies, pursuant to 18 U.S.C. Section 1350, that:
(1) The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
(2) The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
Date: February 26, 2026
/s/RICHARD S. LINDAHL
Richard S. Lindahl
Chief Financial Officer

Corporate Policy Emergent BioSolutions Compensation Recovery Policy 1 Corporate Policy : Emergent BioSolutions Compensation Recovery Policy Emergent BioSolutions Compensation Recovery Policy Purpose & Scope The purpose of this Compensation Recovery Policy (the “Policy”), is to: • Comply with applicable law and any rules or regulations adopted by the Securities and Exchange Commission, including Section 10D-1 under the Securities Exchange Act of 1934, the rules promulgated by the Securities and Exchange Commission thereunder, and the listing standards of the New York Stock Exchange (collectively, the “Applicable Rules”). • Describe the specific events that trigger a clawback of Excess Compensation from Executive Officers (as each term is defined herein). This Policy’s scope: • This Policy applies to all Executive Officers and is applicable to all Incentive-Based Compensation received by Executive Officers after October 26, 2023 (the “Effective Date”). • Each Executive Officer shall be required to execute the acknowledgement in Appendix A of this Policy as soon as practicable after the later of (i) the Effective Date and (ii) the date on which the employee is designated as an Executive Officer; provided, however, that failure to execute such acknowledgement shall have no impact on the enforceability of this Policy. Policy Owner: Jessica Perl Policy Sponsor: Jessica Perl Policy Contact: perlj@ebsi.com Last updated: 29 Oct 2025

Corporate Policy Emergent BioSolutions Compensation Recovery Policy 2 Policy Emergent will comply with the Applicable Rules and take action to recoup Excess Compensation from current and former Executive Officers. 1. Clawback In the event the Company is required to prepare an Accounting Restatement, any Executive Officer who received Excess Compensation during the Look-back Period will be required to repay or forfeit any such Excess Compensation reasonably promptly. For purposes of this Policy, the date the Company is required to prepare an Accounting Restatement is deemed to be the earlier of the date (i) the Board concludes, or reasonably should have concluded, that the Company is required to prepare an Accounting Restatement, or (ii) a court, regulator, or other legally authorized body directs the Company to prepare an Accounting Restatement. Recovery of erroneously received compensation under this Policy is on a “no fault” basis, meaning that it will occur regardless of whether the Executive Officer engaged in misconduct or was otherwise directly or indirectly responsible, in whole or in part, for the Accounting Restatement. In the event of misconduct under this section, the Company’s ability to recover compensation will not be limited to Excess Compensation. No Executive Officer may be indemnified by the Company, or any of its affiliates, from losses arising from the application of this Policy. Any employment contract with any Executive Officer shall be subject to this Policy. 2. Method of Repayment; Conditions for Non-Recovery To the extent permitted by applicable law, including the Applicable Rules, the Board shall seek to recoup Excess Compensation by all legal means available, including, but not limited to, by requiring any affected Executive Officer to repay such amount to the Company in a lump sum or over time, by set-off, by cancelling outstanding awards, by reducing future compensation of the affected Executive Officer, or by such other means or combination of means as the Board, in its sole discretion, determines to be appropriate. At the direction of the Board, the Company shall take all actions reasonable and appropriate to recover Excess Compensation from any applicable Executive Officer, and such Executive Officer shall be required to reimburse the Company for any and all expenses reasonably incurred (including legal fees) by the Company in recovering such Excess Compensation in accordance with this Policy.

Corporate Policy Emergent BioSolutions Compensation Recovery Policy 3 The Board may determine that repayment of Excess Compensation (or a portion thereof) is not required only where it determines that recovery would be impracticable and (i) the direct expense paid to a third party to assist in enforcing this Policy would exceed the amount to be recovered, provided the Company has (A) made a reasonable attempt to recover such Excess Compensation, (B) documented such reasonable attempt, and (C) provided such documentation to the New York Stock Exchange, or (ii) recovery would likely cause an otherwise tax-qualified retirement plan, under which benefits are broadly available to employees, to fail to meet the requirements of 26 U.S.C. 401(a)(13) or 26 U.S.C. 411(a) and the regulations thereunder. Any forfeiture or recoupment under this Policy will be in addition to any other remedies that may be available under applicable law or Company policy, plan or agreement, including termination of employment or institution of civil proceedings. Any determination regarding this Policy and any application and implementation thereof need not be uniform with respect to each Executive Officer or payment recovered or forfeited under this Policy. Any determination made by the Board under this Policy will be final and binding on all affected individuals. 3. Disclosure; Reporting The Company shall disclose this Policy in accordance with the Applicable Rules Act and shall report any triggering event in accordance with the requirements of Applicable Rules. Role & Responsibilities Role Responsibilities Legal Maintain and implement Policy. Board of Directors of the Company Administer the Policy (unless the Board designates the Compensation Committee of the Board (the “Committee”) as the administrator of the Policy, in which case references to the Board will be deemed to be references to the Committee). Definitions • Accounting Restatement: An accounting restatement due to the material noncompliance of the Company with any financial reporting requirement under securities laws, including any required accounting restatement to correct an error in previously issued financial statements that is material to the previously issued financial statements, or that corrects an error that is not material to previously issued financial statements but would result in a material misstatement if the error were corrected in the current period or left uncorrected in the current period. Changes to financial

Corporate Policy Emergent BioSolutions Compensation Recovery Policy 4 statements that do not constitute an Accounting Restatement include retroactive: (i) application of a change from one generally accepted accounting principle to another generally accepted accounting principle; (ii) revisions to reportable segment information due to a change in internal organization; (iii) reclassification due to a discontinued operation; (iv) application of a change in reporting entity, such as from a reorganization of entities under common control; and (v) revisions for stock splits, reverse stock splits, stock dividends, or other changes in capital structure. • Board: The Board of Directors of the Company. • Company: Emergent BioSolutions Inc. • Excess Compensation: Any amount of Incentive-Based Compensation received by an Executive Officer after commencement of service as an Executive Officer that exceeds the amount of Incentive-Based Compensation that otherwise would have been received had it been determined based on the Accounting Restatement, computed without regard to any taxes paid. For Incentive Compensation based on stock price or total shareholder return, where the amount to be recovered is not subject to mathematical recalculation directly from information in the Accounting Restatement, the amount to be recovered shall be based on a reasonable estimate of the effect of the Accounting Restatement on the stock price or total shareholder return, as applicable, and the Corporation shall retain documentation of the determination of such estimate and provide such documentation to NYSE if so required by the Applicable Rules. Incentive-Based Compensation is deemed received during the fiscal year during which the applicable financial reporting measure, stock price and/or total shareholder return measure, upon which the payment is based, is achieved, even if the grant or payment occurs after the end of such period. • Exchange Act: Securities Exchange Act of 1934, as amended. • Executive Officer: An individual who is, or was during the Look-back Period, an executive officer of the Company within the meaning of Rule 10D-1(d) under the Exchange Act. • Incentive-Based Compensation: Any compensation that is granted, earned or vested based wholly or in part on stock price, total shareholder return, and/or the attainment of (i) any financial reporting measure(s) that are determined and presented in accordance with the accounting principles used in preparing the Company’s financial statements and/or (ii) any other measures that are derived in whole or in part from such measures. Compensation that does not constitute “Incentive-Based Compensation” includes equity incentive awards for which the grant is not contingent upon achieving any financial reporting measure performance goal for an individual to receive such award and that vest exclusively upon completion of a specified employment period, without any performance condition, and bonus awards that are discretionary or based on subjective goals or goals unrelated to financial reporting measures. • Look-back Period: The three completed fiscal years preceding the date the Company is required to prepare an Accounting Restatement.

Corporate Policy Emergent BioSolutions Compensation Recovery Policy 5 References and Supporting Documents • Rule 10D-1 under the Exchange Act • NYSE Listed Company Manual Rule 303A.14 • Section 16 of the Exchange Act • Section 304 of the Sarbanes-Oxley Act of 2002 • Section 954 of the Dodd-Frank Wall Street Reform and Consumer Protection Act Effective Date: October 26, 2023