UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-Q
| (Mark One) | ||||||||
| QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | ||||||||
| For the quarterly period ended | ||||||||
| OR | ||||||||
| TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | ||||||||
| For the transition period from to | ||||||||
Commission file number: 001-33137

(Exact Name of Registrant as Specified in Its Charter)
| (State or Other Jurisdiction of Incorporation or Organization) | (I.R.S. Employer Identification No.) | |||||||
| (Address and zip code of Principal Executive Offices) | ||||||||
(240 ) 631-3200
(Registrant's Telephone Number, Including Area Code)
| Securities registered pursuant to Section 12(b) of the Act | ||||||||
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||||||
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ☒ Yes ☐ No
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). ☒ Yes ☐ No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of "large accelerated filer," "accelerated filer," "smaller reporting company," and "emerging growth company" in Rule 12b-2 of the Exchange Act. (Check one):
| ☒ | Accelerated filer | ☐ | |||||||||
| Non-accelerated filer | ☐ | Smaller reporting company | |||||||||
| Emerging growth company | |||||||||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
As of October 29, 2021 the registrant had 53,798,664 shares of common stock outstanding.
1
Emergent BioSolutions Inc.
Index to Form 10-Q
| Page No. | ||||||||
2
EMERGENT BIOSOLUTIONS INC.
PART I. FINANCIAL INFORMATION
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This quarterly report on Form 10-Q and the documents we incorporate by reference include forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. All statements, other than statements of historical fact, including statements regarding the future earnings and performance of Emergent BioSolutions Inc. or any of our businesses, our strategy, future operations, future financial position, future revenues, projected costs, prospects, plans and objectives of management and the continued impact of the COVID-19 pandemic, are forward-looking statements. We generally identify forward-looking statements by using words like "will," "believes," "expects," "anticipates," "intends," "plans," "forecasts," "estimates" and similar expressions in conjunction with, among other things, discussions of financial performance or financial condition, growth strategy, product sales, manufacturing capabilities, product development and regulatory approvals or expenditures. These forward-looking statements are based on our current intentions, beliefs and expectations regarding future events. We cannot guarantee that any forward-looking statement will be accurate. You should realize that if underlying assumptions prove inaccurate or unknown risks or uncertainties materialize, actual results could differ materially from our expectations. You are, therefore, cautioned not to place undue reliance on any forward-looking statement. Any forward-looking statement speaks only as of the date on which such statement is made, and, except as required by law, we do not undertake to update any forward-looking statement to reflect new information, events or circumstances.
There are a number of important factors that could cause our actual results to differ materially from those indicated by such forward-looking statements, including, among others:
▪the availability of U.S. Government ("USG") funding for procurement of AV7909 and/or BioThrax or ACAM2000 and our other USG procurement and development contracts;
▪the timing of our submission of an application for and our ability to secure licensure of AV7909 from the U.S. Food and Drug Administration ("FDA") within the anticipated timeframe, if at all;
▪our ability to perform under our contracts with the USG, including the timing of and specifications relating to deliveries;
▪our ability to meet our commitments to continued quality and manufacturing compliance at our manufacturing facilities, and the potential impact on our ability to continue production of bulk drug substance for Johnson & Johnson’s COVID-19 vaccine;
▪our ability to provide contract development and manufacturing ("CDMO") services for the development and/or manufacture of product candidates of our customers at required levels and on required timelines;
▪our ability and the ability of our contractors and suppliers to maintain compliance with current good manufacturing practices and other regulatory obligations;
▪our ability to obtain and maintain regulatory approvals for our product candidates and the timing of any such approvals;
▪our ability to negotiate additional USG procurement or follow-on contracts for our Public Health Threat ("PHT") products that have expired or will be expiring;
▪the negotiation of further commitments or contracts related to the collaboration and deployment of capacity toward future commercial manufacturing under our CDMO contracts;
▪our ability to successfully appeal the patent litigation decision related to NARCAN® (naloxone hydrochloride) Nasal Spray 4mg/spray, and the impact of competition from potential generic and branded naloxone entrants on NARCAN® Nasal Spray;
▪the results of pending shareholder litigation and potential impact on our business;
▪our ability to develop a safe and effective treatment for COVID-19 and obtain authorization for emergency use for or approval of such treatment from the FDA;
▪our ability to comply with the operating and financial covenants required by our senior secured credit facilities (Senior Secured Credit Facilities) and our 3.875% Senior Unsecured Notes due 2028;
▪the procurement of products by USG entities under regulatory exemptions prior to approval by the FDA and corresponding procurement by government entities outside of the United States under regulatory exemptions prior to approval by the corresponding regulatory authorities in the applicable country;
▪the full impact of COVID-19 disease ("COVID-19") on our markets, operations and employees as well as those of our customers and suppliers;
▪the impact on our revenues from and duration of declines in sales of our vaccine products that target travelers due to the reduction of international travel caused by the COVID-19 pandemic;
3
EMERGENT BIOSOLUTIONS INC.
▪our ability to identify and acquire companies, businesses, products or product candidates that satisfy our selection criteria;
▪the success of our commercialization, marketing and manufacturing capabilities and strategy; and
▪the accuracy of our estimates regarding future revenues, expenses, capital requirements and needs for additional financing.
The foregoing sets forth many, but not all, of the factors that could cause actual results to differ from our expectations in any forward-looking statement. New factors emerge from time to time and it is not possible for management to predict all such factors, nor can it assess the impact of any such factor on the business or the extent to which any factor, or combination of factors, may cause results to differ materially from those contained in any forward-looking statement. You should consider this cautionary statement, the risk factors identified in the section entitled “Risk Factors” in this quarterly report on Form 10-Q and the risk factors identified in our other periodic reports filed with the Securities and Exchange Commission ("SEC") when evaluating our forward-looking statements.
NOTE REGARDING COMPANY REFERENCES
References in this report to “Emergent,” the “Company,” “we,” “us,” and “our” refer to Emergent BioSolutions Inc. and its consolidated subsidiaries.
NOTE REGARDING TRADENAMES
BioThrax® (Anthrax Vaccine Adsorbed), RSDL® (Reactive Skin Decontamination Lotion Kit), BAT® (Botulism Antitoxin Heptavalent (A,B,C,D,E,F,G)-(Equine)), Anthrasil® (Anthrax Immune Globulin Intravenous (Human)), VIGIV (Vaccinia Immune Globulin Intravenous (Human)), Trobigard® (atropine sulfate, obidoxime chloride), ACAM2000® (Smallpox (Vaccinia) Vaccine, Live), Vivotif® (Typhoid Vaccine Live Oral Ty21a), Vaxchora® (Cholera Vaccine, Live, Oral), NARCAN® (naloxone HCI) Nasal Spray and any and all Emergent brands, products, services and feature names, logos and slogans are trademarks or registered trademarks of Emergent or its subsidiaries in the United States or other countries. All other brands, products, services and feature names or trademarks are the property of their respective owners.
4
ITEM 1. FINANCIAL STATEMENTS
Emergent BioSolutions Inc.
Condensed Consolidated Balance Sheets
(unaudited, in millions, except per share amounts)
| September 30, 2021 | December 31, 2020 | ||||||||||
| ASSETS | |||||||||||
| Current assets: | |||||||||||
| Cash and cash equivalents | $ | $ | |||||||||
| Restricted cash | |||||||||||
| Accounts receivable, net | |||||||||||
| Inventories, net | |||||||||||
| Prepaid expenses and other current assets | |||||||||||
| Total current assets | |||||||||||
| Property, plant and equipment, net | |||||||||||
| Intangible assets, net | |||||||||||
| Goodwill | |||||||||||
| Other assets | |||||||||||
| Total assets | $ | $ | |||||||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | |||||||||||
| Current liabilities: | |||||||||||
| Accounts payable | $ | $ | |||||||||
| Accrued expenses | |||||||||||
| Accrued compensation | |||||||||||
| Debt, current portion | |||||||||||
| Other current liabilities | |||||||||||
| Total current liabilities | |||||||||||
| Contingent consideration, net of current portion | |||||||||||
| Debt, net of current portion | |||||||||||
| Deferred tax liability | |||||||||||
| Contract liabilities, net of current portion | |||||||||||
| Other liabilities | |||||||||||
| Total liabilities | |||||||||||
| Stockholders' equity: | |||||||||||
Preferred stock, $ | |||||||||||
Common stock, $ | |||||||||||
| Additional paid-in capital | |||||||||||
Treasury stock, at cost, | ( | ( | |||||||||
| Accumulated other comprehensive loss, net | ( | ( | |||||||||
| Retained earnings | |||||||||||
| Total stockholders' equity | |||||||||||
| Total liabilities and stockholders' equity | $ | $ | |||||||||
See accompanying notes.
5
Emergent BioSolutions Inc.
Condensed Consolidated Statements of Operations
(unaudited, in millions, except per share amounts)
| Three Months Ended September 30, | Nine Months Ended September 30, | ||||||||||||||||||||||
| 2021 | 2020 | 2021 | 2020 | ||||||||||||||||||||
| Revenues: | |||||||||||||||||||||||
| Product sales, net | $ | $ | $ | $ | |||||||||||||||||||
| Contract development and manufacturing: | |||||||||||||||||||||||
| Services | |||||||||||||||||||||||
| Leases | ( | ||||||||||||||||||||||
| Total contract development and manufacturing | |||||||||||||||||||||||
| Contracts and grants | |||||||||||||||||||||||
| Total revenues | |||||||||||||||||||||||
| Operating expenses: | |||||||||||||||||||||||
| Cost of product sales | |||||||||||||||||||||||
| Cost of contract development and manufacturing | |||||||||||||||||||||||
| Research and development | |||||||||||||||||||||||
| Selling, general and administrative | |||||||||||||||||||||||
| Amortization of intangible assets | |||||||||||||||||||||||
| Total operating expenses | |||||||||||||||||||||||
| Income (loss) from operations | ( | ||||||||||||||||||||||
| Other income (expense): | |||||||||||||||||||||||
| Interest expense | ( | ( | ( | ( | |||||||||||||||||||
| Other, net | ( | ( | |||||||||||||||||||||
| Total other income (expense), net | ( | ( | ( | ( | |||||||||||||||||||
| Income (loss) before income taxes | ( | ||||||||||||||||||||||
| Income taxes | ( | ( | ( | ||||||||||||||||||||
| Net income (loss) | $ | ( | $ | $ | $ | ||||||||||||||||||
| Net income (loss) per common share | |||||||||||||||||||||||
| Basic | $ | ( | $ | $ | $ | ||||||||||||||||||
| Diluted | $ | ( | $ | $ | $ | ||||||||||||||||||
| Shares used in computing income (loss) per share | |||||||||||||||||||||||
| Basic | |||||||||||||||||||||||
| Diluted | |||||||||||||||||||||||
See accompanying notes.
6
Emergent BioSolutions Inc.
Condensed Consolidated Statements of Comprehensive Income
(unaudited, in millions)
| Three Months Ended September 30, | Nine Months Ended September 30, | ||||||||||||||||||||||
| 2021 | 2020 | 2021 | 2020 | ||||||||||||||||||||
| Net income (loss) | $ | ( | $ | $ | $ | ||||||||||||||||||
| Other comprehensive income (loss), net of tax: | |||||||||||||||||||||||
| Foreign currency translation | ( | ( | ( | ||||||||||||||||||||
| Unrealized gains (losses) on hedging activities | ( | ( | |||||||||||||||||||||
| Total other comprehensive income (loss) | ( | ( | |||||||||||||||||||||
| Comprehensive income (loss) | $ | ( | $ | $ | $ | ||||||||||||||||||
See accompanying notes.
7
Emergent BioSolutions Inc.
Condensed Consolidated Statements of Cash Flows
(unaudited, in millions)
| Nine Months Ended September 30, | |||||||||||
| 2021 | 2020 | ||||||||||
| Cash flows (used in) provided by operating activities: | |||||||||||
| Net income | $ | $ | |||||||||
| Adjustments to reconcile net income to net cash (used in) provided by operating activities: | |||||||||||
| Share-based compensation expense | |||||||||||
| Depreciation and amortization | |||||||||||
| Adjustment for prior period lease receivables (Note 10) | |||||||||||
| Change in fair value of contingent consideration, net | |||||||||||
| Amortization of deferred financing costs | |||||||||||
| Deferred income taxes | ( | ||||||||||
| Impairment of IPR&D | |||||||||||
| Other | |||||||||||
| Changes in operating assets and liabilities: | |||||||||||
| Accounts receivable | ( | ||||||||||
| Inventories | ( | ( | |||||||||
| Prepaid expenses and other assets | ( | ( | |||||||||
| Accounts payable | |||||||||||
| Accrued expenses and other liabilities | ( | ||||||||||
| Accrued compensation | ( | ||||||||||
| Contract liabilities | ( | ( | |||||||||
| Net cash (used in) provided by operating activities: | ( | ||||||||||
| Cash flows used in investing activities: | |||||||||||
| Purchases of property, plant and equipment | ( | ( | |||||||||
| Milestone payment from prior asset acquisition | ( | ||||||||||
| Net cash used in investing activities: | ( | ( | |||||||||
| Cash flows (used in) provided by financing activities: | |||||||||||
| Principal payments on revolving credit facility | ( | ||||||||||
| Principal payments on term loan facility | ( | ( | |||||||||
| Principal payments on convertible senior notes | ( | ||||||||||
| Proceeds from senior unsecured notes | |||||||||||
| Proceeds from share-based compensation activity | |||||||||||
| Debt issuance costs | ( | ||||||||||
| Taxes paid for share-based compensation activity | ( | ( | |||||||||
| Contingent consideration payments | ( | ( | |||||||||
| Net cash (used in) provided by financing activities: | ( | ||||||||||
| Effect of exchange rate changes on cash, cash equivalents and restricted cash | ( | ( | |||||||||
| Net change in cash, cash equivalents and restricted cash | ( | ||||||||||
| Cash, cash equivalents and restricted cash at beginning of period | |||||||||||
| Cash, cash equivalents and restricted cash at end of period | $ | $ | |||||||||
| Supplemental disclosure of cash flow information: | |||||||||||
| Cash paid during the period for interest | $ | $ | |||||||||
| Cash paid during the period for income taxes | $ | $ | |||||||||
| Supplemental information on non-cash investing and financing activities: | |||||||||||
| Purchases of property, plant and equipment unpaid at period end | $ | $ | |||||||||
Reconciliation of cash and cash equivalent and restricted cash at September 30, 2021 and December 31, 2020: | |||||||||||
| Cash and cash equivalents | $ | $ | |||||||||
| Restricted cash | |||||||||||
| Total | $ | $ | |||||||||
See accompanying notes.
8
Emergent BioSolutions Inc.
Condensed Consolidated Statements of Changes in Stockholders' Equity
(unaudited, in millions)
$ | Additional Paid-In Capital | Treasury Stock | Accumulated Other Comprehensive Loss | Retained Earnings | Total Stockholders' Equity | |||||||||||||||||||||||||||||||||||||||||||||
Shares | Amount | Shares | Amount | |||||||||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2020 | $ | $ | ( | $ | ( | $ | ( | $ | $ | |||||||||||||||||||||||||||||||||||||||||
| Share-based compensation activity | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||
| Net income (loss) | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||
| Other comprehensive income (loss) | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||
| Balance at September 30, 2021 | $ | $ | ( | $ | ( | $ | ( | $ | $ | |||||||||||||||||||||||||||||||||||||||||
| Balance at June 30, 2021 | $ | $ | ( | $ | ( | $ | ( | $ | $ | |||||||||||||||||||||||||||||||||||||||||
| Share-based compensation activity | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||
| Net income (loss) | — | — | — | — | — | — | ( | ( | ||||||||||||||||||||||||||||||||||||||||||
| Other comprehensive income (loss) | — | — | — | — | — | ( | — | ( | ||||||||||||||||||||||||||||||||||||||||||
| Balance at September 30, 2021 | $ | $ | ( | $ | ( | $ | ( | $ | $ | |||||||||||||||||||||||||||||||||||||||||
| Balance at December 31, 2019 | $ | $ | ( | $ | ( | $ | ( | $ | $ | |||||||||||||||||||||||||||||||||||||||||
| Share-based compensation activity | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||
| Net income (loss) | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||
| Other comprehensive income (loss) | — | — | — | — | — | ( | — | ( | ||||||||||||||||||||||||||||||||||||||||||
| Balance at September 30, 2020 | $ | $ | ( | $ | ( | $ | ( | $ | $ | |||||||||||||||||||||||||||||||||||||||||
| Balance at June 30, 2020 | $ | $ | ( | $ | ( | $ | ( | $ | $ | |||||||||||||||||||||||||||||||||||||||||
| Share-based compensation activity | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||
| Net income (loss) | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||
| Other comprehensive income (loss) | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||
| Balance at September 30, 2020 | $ | $ | ( | $ | ( | $ | ( | $ | $ | |||||||||||||||||||||||||||||||||||||||||
See accompanying notes.
9
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
1. Business
Organization and business
Emergent BioSolutions Inc. (the "Company" or "Emergent") is a global life sciences company focused on providing civilian and military populations with a portfolio of innovative preparedness and response products and solutions that address accidental, deliberate and naturally occurring public health threats ("PHTs," each a “PHT”).
The Company is focused on the following five distinct PHT categories: Chemical, Biological, Radiological, Nuclear and Explosives ("CBRNE"); emerging infectious diseases ("EID"); travel health; emerging health crises; acute/emergency care; and contract development and manufacturing ("CDMO"). The Company has a product portfolio of ten products (vaccines, therapeutics, and drug-device combination products) that contribute a substantial portion of our revenue. The Company has two product candidates that are procured under special circumstances by certain government agencies, although they are not approved by the U.S. Food and Drug Administration ("FDA"). The U.S. government (the "USG") is the Company's largest customer and provides the Company with substantial funding for the development of a number of its product candidates.
The Company's product and services portfolio includes:
Vaccines
▪ACAM2000® (Smallpox (Vaccinia) Vaccine, Live), the only single-dose smallpox vaccine licensed by the FDA for active immunization against smallpox disease for persons determined to be at high risk for smallpox infection;
▪BioThrax® (Anthrax Vaccine Adsorbed), the only vaccine licensed by the FDA, for the general use prophylaxis and post-exposure prophylaxis of anthrax disease;
▪Vaxchora® (Cholera Vaccine, Live, Oral), the only single-dose oral vaccine licensed by the FDA and the European Medicines Agency ("EMA") for the prevention of cholera; and
▪Vivotif® (Typhoid Vaccine Live Oral Ty21a), the only oral vaccine licensed by the FDA for the prevention of typhoid fever.
Devices
▪NARCAN® (naloxone HCl) Nasal Spray, the first needle-free formulation of naloxone approved by the FDA and Health Canada, for the emergency treatment of known or suspected opioid overdose as manifested by respiratory and/or central nervous system depression; and
▪RSDL® (Reactive Skin Decontamination Lotion Kit), the only medical device cleared by the FDA to remove or neutralize the following chemical warfare agents from the skin: tabun, sarin, soman, cyclohexyl sarin, VR, VX, mustard gas and T-2 toxin.
Therapeutics
▪raxibacumab (Anthrax Monoclonal), a fully human monoclonal antibody therapeutic licensed by the FDA for the treatment and prophylaxis of inhalational anthrax;
▪Anthrasil® (Anthrax Immune Globulin Intravenous (Human)), the only polyclonal antibody therapeutic licensed by the FDA and Health Canada for the treatment of inhalational anthrax;
▪BAT® (Botulism Antitoxin Heptavalent (A,B,C,D,E,F,G)-(Equine)), the only heptavalent antibody therapeutic licensed by the FDA and Health Canada for the treatment of botulism; and;
▪VIGIV (Vaccinia Immune Globulin Intravenous (Human)), the only polyclonal antibody therapeutic licensed by the FDA and Health Canada to address certain complications from smallpox vaccination.
Procured Product Candidates
•AV7909® (Anthrax Vaccine Absorbed with Adjuvant), is a procured product candidate being developed as a next generation anthrax vaccine for post-exposure prophylaxis of disease resulting from suspected or confirmed Bacillus anthracis exposure. The USG has largely switched from procuring BioThrax to AV7909 for the Strategic National Stockpile ("SNS") prior to its approval by the FDA; and
10
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
•Trobigard® is a combination drug-device auto-injector procured product candidate that contains atropine sulfate and obidoxime chloride. It has not been approved by the FDA, but it is procured by certain authorized government buyers under special circumstances for potential use as a nerve agent countermeasure.
Contract Development and Manufacturing Services
The Company's contract development and manufacturing ("CDMO") service offerings cover development services, drug substance manufacturing, drug product manufacturing, and when necessary, suite reservations, which depending on facts and circumstances could be considered a lease. These services are provided across the pharmaceutical and biotechnology industries as well as the USG and non-governmental organizations. The Company's technology platforms include mammalian, microbial, viral, plasma and advanced therapies utilizing our core capabilities for manufacturing to third parties on a clinical and commercial (small and large) scale. Additional services include fill/finish formulation and analytical development services for injectable and other sterile products, inclusive of process design, technical transfer, manufacturing validations, aseptic filling, lyophilization, final packaging and stability studies, as well as manufacturing of vial and pre-filled syringe formats on multiple platforms.
The Company operates as one operating segment.
2. Basis of Presentation and Principles of Consolidation
Basis of presentation
Significant accounting policies
During the nine months ended September 30, 2021, there have been no significant changes to the Company's summary of significant accounting policies contained in the Company's Annual Report on Form 10-K for the year ended December 31, 2020, as filed with the SEC that have materially impacted the presentation of the Company's financial statements. During the period, the Company has adjusted its CDMO revenue recognition policy, which resulted in immaterial out-of-period adjustments which are further discussed below. The Company's adjusted CDMO revenue recognition policies are as follows:
The Company performs CDMO services for third parties. Under these contracts, activities can include drug substance and drug product manufacturing services for injectable and other sterile products, and development services such as pharmaceutical product process development, process design, technology transfer, manufacturing validations, laboratory analytical development support, aseptic filling, lyophilization, final packaging, stability studies, and suite-reservations. These contracts vary in duration, activities, and number of performance obligations. Performance obligations identified under these arrangements may include drug substance and/or drug product manufacturing, technology transfer activities, and suite-reservations.
Drug substance and drug product manufacturing performance obligations are recognized as revenue over-time because the Company’s performance does not create an asset with an alternative use and the Company has an enforceable right to payment for performance completed as work is performed. In drug product arrangements, the customer typically owns and supplies the active pharmaceutical ingredient, or API, that is used in the manufacturing process; in drug substance arrangements, the customer provides certain seed material that is used in the manufacturing process. The transaction price is stated in the agreement as a fixed price per unit, with no contractual provision for a refund or price concession. We use an input method to measure progress toward the satisfaction of
11
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
the related performance obligations based on costs incurred as a percentage of total costs to complete which we believe best depicts the transfer of control of goods or services promised to our customers.
For arrangements with development services or where we have identified technology transfer activities to be a separate performance obligation, revenues are recognized over-time as the service is provided as there is no alternative future use to the Company for the asset created and the Company has an enforceable right to payment for performance completed as of that date. We use an input method to measure progress toward the satisfaction of the related performance obligations based on costs incurred as a percentage of total costs to complete which we believe best depicts the transfer of control of goods or services promised to our customers.
Suite reservations are classified as leases when the customer directs the use of the identified suite and obtains substantially all the economic benefits from the manufacturing capacity. If a customer reserves more than one suite, the allocation of contract value is based on relative selling price which varies due to size, location, capacity, production capability for drug product or drug substance, and the time of planned use. The associated revenue is recognized on a straight-line basis over the period of performance. For arrangements that contain both lease and non-lease components, consideration in the contract is allocated on a relative standalone selling price basis.
The Company’s CDMO customer contracts generally include provisions entitling the Company to a termination penalty when the contract is terminated prior to the contract’s nominal end date. The termination penalties in the customer contracts vary but are generally considered substantive for accounting purposes and create enforceable rights and obligations throughout the stated duration of the contract. The Company accounts for a contract cancellation as a contract modification. The determination of the contract termination penalty is based on the terms stated in the related customer agreement. As of the modification date, the Company updates its estimate of the transaction price, subject to constraints, and recognizes the amount over the remaining performance period or measure of progress under the arrangement.
Fair value measurements
As of September 30, 2021 and December 31, 2020, the Company had no other significant assets or liabilities that were measured at fair value.
12
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
Out-of-period adjustments
During the three months ended September 30, 2021, the Company identified and recorded immaterial out-of-period adjustments. Historically, the Company has recognized revenue for drug substance and drug product manufacturing performance obligations when the goods have been released, legal title has passed and the goods are in the customer's possession. Beginning with the current period, the Company will recognize revenue over time using an input measure based on costs incurred as a percentage of total estimated contract costs to recognize batch production and fill-finish revenue. As batch production and fill-finish manufacturing generally take place over short intervals, the adjustments to the financial statements were not material. Additionally, the Company determined that the classification of its suite reservations, when the customer directs the use of the identified suite and obtains substantially all the economic benefits reflected in CDMO service revenue, are more appropriately classified as leases. Although either classification generally results in recognition of revenue on a straight line basis over-time, the Company identified one lease component commencement date change which impacted the revenue recognized during our 2020 and 2021 periods. The Company has also included incremental lease accounting disclosures in these financial statements (see Note 10).
The Company evaluated the materiality of the out-of-period adjustments from quantitative and qualitative perspectives and concluded that the adjustments were immaterial to the Company’s prior period interim and annual consolidated financial statements. As a result, no amendments to previously filed interim or annual periodic reports are required. These adjustments resulted in the following out-of-period adjustments:
| Three Months Ended September 30, 2021 | Nine Months Ended September 30, 2021 | ||||||||||
| Contract development and manufacturing revenue: | |||||||||||
| Services | |||||||||||
| Leases | ( | ( | |||||||||
| Total contract development and manufacturing revenue | |||||||||||
| Cost of CDMO | |||||||||||
| Income before income taxes | |||||||||||
| Net income | |||||||||||
The condensed consolidated statements of operations and statements of changes in stockholders' equity for the three and nine months ended September 30, 2021, the condensed consolidated balance sheet as of September 30, 2021 and the condensed consolidated statements of cash flows for the nine months ended September 30, 2021 reflect the above adjustments.
In addition, during the three and nine months ended September 30, 2021, the Company revised its presentation on the condensed consolidated statement of operations to separately present (i) lease revenues as opposed to combining with CDMO services revenues as the Company had previously reported and (ii) cost of contract development and manufacturing as opposed to combining with cost of product sales. As the Company's lease revenue is solely associated with CDMO services and is substantially related to one arrangement which will not continue after 2021, the Company has combined the costs of CDMO services and leases within the condensed consolidated statement of operations. All associated prior period amounts have been reclassified to conform to the current period presentation.
Recently issued accounting standards
Recently Adopted
ASU 2019-12, Simplifications to Accounting for Income Taxes ("ASU 2019-12")
In December 2019, the FASB issued ASU 2019-12. ASU 2019-12 removes certain exceptions for recognizing deferred taxes for investments, performing intra-period allocation and calculating income taxes in interim periods. The ASU also adds guidance to reduce complexity in certain areas, including deferred taxes for goodwill and allocating taxes for members of a consolidated group. ASU 2019-12 is effective for all entities for fiscal years beginning after December 15, 2020, and earlier adoption is permitted. As of January 1, 2021, the Company adopted the standard, which did not have a material impact on the Company's consolidated financial statements.
13
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
Not Yet Adopted
ASU 2020-04, Reference Rate Reform (Topic 848): Facilitation of the Effects of Reference Rate Reform on Financial Reporting
3. Inventories, net
The components of inventory are as follows:
| September 30, 2021 | December 31, 2020 | ||||||||||
| Raw materials and supplies | $ | $ | |||||||||
| Work-in-process | |||||||||||
| Finished goods | |||||||||||
| Total inventories, net | $ | $ | |||||||||
Inventories, net is stated at the lower of cost or net realizable value. During the nine months ended September 30, 2021, the Company recorded inventory write-offs at its Bayview facility of $41.5 million, which were directly or indirectly the result of the cross-contamination event at the Bayview facility identified during the three months ended June 30, 2021. The inventory write-off resulted from the Company's plan to discard raw materials and in-process batches that were deemed unusable. The charge was reflected as a component of cost of contract development and manufacturing.
4. Property, plant and equipment, net
Property, plant and equipment, net consisted of the following:
| September 30, 2021 | December 31, 2020 | ||||||||||
| Land and improvements | $ | $ | |||||||||
| Buildings, building improvements and leasehold improvements | |||||||||||
| Furniture and equipment | |||||||||||
| Software | |||||||||||
| Construction-in-progress | |||||||||||
| Property, plant and equipment, gross | |||||||||||
| Accumulated depreciation | ( | ( | |||||||||
| Total property, plant and equipment, net | $ | $ | |||||||||
As of September 30, 2021 and December 31, 2020, construction-in-progress primarily includes costs incurred related to construction to advance the Company's CDMO capabilities. These costs include capital expenditures related to our Biomedical Advanced Research and Development Authority ("BARDA") COVID-19 Development Public Private Partnership (see Note 10).
14
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
5. Leases
The Company is the lessee for operating leases for corporate offices, research and development facilities and manufacturing facilities. We determine if an arrangement is a lease at inception. Operating leases are included in right-of-use ("ROU") assets and liabilities. (For a discussion of lessor activities, see Note 10.)
The components of lease expense were as follows:
| Three Months Ended September 30, | Nine Months Ended September 30, | ||||||||||||||||||||||
| 2021 | 2020 | 2021 | 2020 | ||||||||||||||||||||
| Operating lease cost: | |||||||||||||||||||||||
| Amortization of right-of-use assets | $ | $ | $ | $ | |||||||||||||||||||
| Interest on lease liabilities | |||||||||||||||||||||||
| Total operating lease cost | $ | $ | $ | $ | |||||||||||||||||||
Operating lease costs are reflected as components of cost of product sales, cost of contract development and manufacturing, research and development expense and selling, general and administrative expense.
Supplemental balance sheet information related to leases was as follows:
| (In millions, except lease term and discount rate) | Balance Sheet location | September 30, 2021 | December 31, 2020 | ||||||||||||||
| Operating lease right-of-use assets | $ | $ | |||||||||||||||
| Operating lease liabilities, current portion | |||||||||||||||||
| Operating lease liabilities | |||||||||||||||||
| Total operating lease liabilities | $ | $ | |||||||||||||||
| Operating leases: | |||||||||||||||||
| Weighted average remaining lease term (years) | |||||||||||||||||
| Weighted average discount rate | % | % | |||||||||||||||
6. Intangible assets
The Company's intangible assets consist of products acquired via business combinations or asset acquisitions. The following tables summarize the Company's intangible assets for the periods ended September 30, 2021 and December 31, 2020:
September 30, 2021 | December 31, 2020 | ||||||||||||||||||||||||||||
| Asset Type | Estimated Life | Cost | Accumulated Amortization | Net | Cost | Accumulated Amortization | Net | ||||||||||||||||||||||
| Products | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||||
| Customer relationships | |||||||||||||||||||||||||||||
| CDMO | |||||||||||||||||||||||||||||
| Total intangible assets | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||||
Amortization expense associated with the Company's intangible assets was recorded as follows:
| Three Months Ended September 30, | Nine Months Ended September 30, | ||||||||||||||||||||||
| 2021 | 2020 | 2021 | 2020 | ||||||||||||||||||||
| Amortization Expense | $ | $ | $ | $ | |||||||||||||||||||
15
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
As of September 30, 2021, the weighted average amortization period remaining for intangible assets was 12.1 years.
The following table provides a roll forward of changes in our goodwill balance:
Goodwill, December 31, 2020 | $ | ||||
| Foreign currency translation | ( | ||||
Goodwill, September 30, 2021 | $ | ||||
7. Fair Value Measurements
The table below presents information about our assets and liabilities that are regularly measured and carried at fair value and indicate the level within the fair value hierarchy of the valuation techniques we utilized to determine fair value:
September 30, 2021 | December 31, 2020 | |||||||||||||||||||||||||
| Total | Level1 | Level 2 | Level 3 | Total | Level1 | Level 2 | Level 3 | |||||||||||||||||||
| Assets: | ||||||||||||||||||||||||||
| Money market accounts | $ | $ | ||||||||||||||||||||||||
| Time deposits | ||||||||||||||||||||||||||
| Total | $ | $ | ||||||||||||||||||||||||
| Liabilities: | ||||||||||||||||||||||||||
| Contingent consideration | $ | $ | ||||||||||||||||||||||||
| Derivative instruments | ||||||||||||||||||||||||||
| Total | $ | $ | ||||||||||||||||||||||||
Contingent Consideration
Contingent consideration liabilities associated with business combinations are measured at fair value. These liabilities represent an obligation of the Company to transfer additional assets to the selling shareholders and owners if future events occur or conditions are met. These liabilities associated with business combinations are measured at fair value at inception and at each subsequent reporting date. The changes in the fair value are primarily due to the expected amount and timing of future net sales, which are inputs that have no observable market. Any changes in fair value for the contingent consideration liabilities related to the Company’s products are classified in the Company's statement of operations as cost of product sales. Any changes in fair value for the contingent consideration liabilities related to the Company’s product candidates are recorded in research and development expense for regulatory and development milestones.
The following table is a reconciliation of the beginning and ending balance of contingent considerations.
| Balance at December 31, 2020 | $ | ||||
| Change in fair value | |||||
| Settlements | ( | ||||
| Balance at September 30, 2021 | $ | ||||
As of September 30, 2021 and December 31, 2020, the current portion of the contingent consideration liability was $32.6 million and $23.9 million, respectively, and was included in other current liabilities on the condensed consolidated balance sheets.
16
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
The recurring Level 3 fair value measurements for the Company's contingent consideration liability include the following significant unobservable inputs:
| Contingent Consideration Liability | Fair Value as of September 30, 2021 | Valuation Technique | Unobservable Input | Range | Weighted Average | ||||||||||||
| Revenue milestone and royalty based | $ | Discounted cash flow | Discount rate | ||||||||||||||
| Probability of payment | |||||||||||||||||
| Projected year of payment | 2021 - 2028 | 2022 | |||||||||||||||
Derivative Instruments
Refer to Note 8, Derivatives, to these condensed consolidated financial statements.
Non-Variable Rate Debt
8. Derivative instruments and hedging activities
Risk management objective of using derivatives
The Company is exposed to certain risks arising from both its business operations and economic conditions. The Company principally manages its exposures to a wide variety of business and operational risks through management of its core business activities. The Company manages economic risks, including interest rate, liquidity, and credit risk primarily by managing the amount, sources, and duration of its assets and liabilities and the use of derivative financial instruments. Specifically, the Company has entered into interest rate swaps to manage exposures that arise from the Company's senior secured credit agreement's payments of variable interest rate debt.
If current fair values of designated interest rate swaps remained static over the next twelve months, the Company would reclassify $5.7 million of net deferred losses from accumulated other comprehensive loss to the condensed consolidated statement of operations over the next twelve month period. All outstanding cash flow hedges mature in October 2023.
Number of Instruments | Notional | ||||||||||
| Interest rate swaps | $ | ||||||||||
| Liability Derivatives | ||||||||||||||
| September 30, 2021 | December 31, 2020 | |||||||||||||
Balance Sheet Location | Fair Value | Balance Sheet Location | Fair Value | |||||||||||
| Interest Rate Swaps | Other Current Liabilities | $ | Other Current Liabilities | $ | ||||||||||
| Other Liabilities | $ | Other Liabilities | $ | |||||||||||
The valuation of the interest rate swaps is determined using widely accepted valuation techniques, including discounted cash flow analysis on the expected cash flows of each interest rate swap. This analysis reflects the
17
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
contractual terms of the interest rate swaps, including the period to maturity, and uses observable market-based inputs, including interest rate curves and implied volatilities. The fair values of interest rate swaps are determined using the market standard methodology of netting the discounted future fixed cash payments (or receipts) and the discounted expected variable cash receipts (or payments). The variable cash payments (or receipts) are based on an expectation of future interest rates (forward curves) derived from observable market interest rate curves. We incorporate credit valuation adjustments in the fair value measurements to appropriately reflect both our own nonperformance risk and the respective counterparty’s nonperformance risk. These credit valuation adjustments were not significant inputs for the fair value calculations for the periods presented. In adjusting the fair value of our derivative contracts for the effect of nonperformance risk, we have considered the impact of netting and any applicable credit enhancements, such as the posting of collateral, thresholds, mutual puts and guarantees. The valuation of interest rate swaps fall into Level 2 in the fair value hierarchy.
| Hedging derivatives | Cumulative Amount of Gain/(Loss) Recognized in OCI on Derivative | Location of Gain or (Loss) Reclassified from Accumulated OCI into Income | Amount of Gain/(Loss) Reclassified from Accumulated OCI into Income | ||||||||||||||
| September 30, | December 31, | Nine Months Ended September 30, | |||||||||||||||
| 2021 | 2020 | 2021 | 2020 | ||||||||||||||
| Interest Rate Swaps | $ | ( | $ | ( | Interest expense | $ | ( | $ | ( | ||||||||
9. Debt
The components of debt are as follows:
| September 30, 2021 | December 31, 2020 | ||||||||||
| Senior secured credit agreement - Term loan due 2023 | $ | $ | |||||||||
| Other | |||||||||||
| Total debt | |||||||||||
| Current portion of long-term debt, net of debt issuance costs | ( | ( | |||||||||
| Unamortized debt issuance costs | ( | ( | |||||||||
| Non-current portion of debt | $ | $ | |||||||||
As of September 30, 2021 and December 31, 2020, debt issuance costs associated with the revolver loan were classified as other current assets and other assets on the Company's consolidated balance sheets because there was no outstanding revolver balance at period end. As of September 30, 2021, the Company had $2.0 million and $2.0 million of debt issuance costs associated with the revolver loan classified as other current assets and other assets, respectively. As of December 31, 2020, the Company had approximately $2.0 million and $3.5 million of debt issuance costs associated with the revolver loan that were classified as other current assets and other assets, respectively.
On August 7, 2020, the Company completed its offering of $450 million aggregate principal amount of 3.875 % Senior Unsecured Notes due 2028 (the “2028 Notes”) of which the majority of the net proceeds were used to pay down the Revolving Credit Facility (as defined below). Interest on the 2028 Notes is payable on February 15th and August 15th of each year until maturity, beginning on February 15, 2021. The 2028 Notes will mature on August 15, 2028.
On or after August 15, 2023, the Company may redeem the 2028 Notes, in whole or in part, at the redemption prices set forth in the related Indenture, plus accrued and unpaid interest. Prior to August 15, 2023 the Company may redeem all or a portion of the 2028 Notes at a redemption price equal to 100 % of the principal amount of the 2028 Notes plus a “make-whole” premium and accrued and unpaid interest. Prior to August 15, 2023, the Company may
18
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
redeem up to 40 % of the aggregate principal amount of the 2028 Notes using the net cash proceeds of certain equity offerings at the redemption price set forth in the related Indenture. Upon the occurrence of a change of control, the Company must offer to repurchase the 2028 Notes at a purchase price of 101 % of the principal amount of such 2028 Notes plus accrued and unpaid interest.
Negative covenants in the Indenture governing the 2028 Notes, among other things, limit the ability of the Company to incur indebtedness and liens, dispose of assets, make investments, enter into certain merger or consolidation transactions and make restricted payments.
Senior secured credit agreement
Also on August 7, 2020, the Company entered into a Second Amendment (the “Credit Agreement Amendment”) to its senior secured credit agreement, dated October 15, 2018, with multiple lending institutions relating to the Company’s senior secured credit facilities (the “Credit Agreement,” and as amended, the “Amended Credit Agreement”), consisting of a senior revolving credit facility (the “Revolving Credit Facility”) and senior term loan facility (the “Term Loan Facility,” and together with the Revolving Credit Facility, the “Senior Secured Credit Facilities”). The Credit Agreement Amendment amended, among other things, the definition of incremental facilities limit, the consolidated net leverage ratio financial covenant by increasing the maximum level, increased the permissible applicable margins based on the Company’s consolidated net leverage ratio and increased the commitment fee that the Company is required to pay in respect of the average daily unused commitments under the Revolving Credit Facility, depending on the Company’s consolidated net leverage ratio.
The Amended Credit Agreement includes (i) a Revolving Credit Facility of $600 million and (ii) a Term Loan Facility with a principal amount of $450 million. The Company may request incremental term loan facilities or increases in the Revolving Credit Facility (each an "Incremental Loan") as long as certain requirements involving our net leverage ratio will be maintained on a pro forma basis. Borrowings under the Revolving Credit Facility and the Term Loan Facility bear interest at a rate per annum equal to (a) a eurocurrency rate plus a margin ranging from 1.25 % to 2.25 % per annum, depending on the Company's consolidated net leverage ratio or (b) a base rate (which is the highest of the prime rate, the federal funds rate plus 0.50 %, and a eurocurrency rate for an interest period of one month plus 1 % plus a margin ranging from 0.25 % to 1.25 %, depending on the Company's consolidated net leverage ratio). The Company is required to make quarterly payments on the last business day of each calendar quarter under the Amended Credit Agreement for accrued and unpaid interest on the outstanding principal balance, based on the above interest rates. In addition, the Company is required to pay commitment fees ranging from 0.15 % to 0.35 % per annum, depending on the Company's consolidated net leverage ratio, for the average daily unused commitments under the Revolving Credit Facility. The Company is to repay the outstanding principal amount of the Term Loan Facility in quarterly installments on the last business day of each calendar quarter based on an annual percentage equal to 2.5 % of the original principal amount of the Term Loan Facility during each of the first two years of the Term Loan Facility, 5 % of the original principal amount of the Term Loan Facility during the third year of the Term Loan Facility and 7.5 % of the original principal amount of the Term Loan Facility during each year of the remainder of the term of the Term Loan Facility until the maturity date of the Term Loan Facility, at which time the entire unpaid principal balance of the Term Loan Facility will be due and payable. The Company has the right to prepay the Term Loan Facility without premium or penalty. The Revolving Credit Facility and the Term Loan Facility mature on October 13, 2023.
The Amended Credit Agreement also requires mandatory prepayments of the Term Loan Facility in the event the Company or its Subsidiaries (a) incur indebtedness not otherwise permitted under the Amended Credit Agreement or (b) receive cash proceeds in excess of $100 million during the term of the Credit Agreement from certain dispositions of property or from casualty events involving their property, subject to certain reinvestment rights. The financial covenants under the Amended Credit Agreement currently require the quarterly presentation of a minimum consolidated 12 -month rolling debt service coverage ratio of 2.50 to 1.00, and a maximum consolidated net leverage ratio of 4.50 to 1.00 (subject to an increase to 5.00 to 1.00 for an applicable four quarter period, at the election of the Company, in connection with a permitted acquisition having an aggregate consideration in excess of $75.0 million). Negative covenants in the Amended Credit Agreement, among other things, limit the ability of the Company to incur indebtedness and liens, dispose of assets, make investments, enter into certain merger or consolidation transactions and make restricted payments. As of the date of these financial statements, the Company is in compliance with all affirmative and negative covenants.
19
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
On January 29, 2014, the Company issued 2.875 % convertible senior notes due 2021 (the "Notes"). The Notes bore interest at a rate of 2.875 % per year, payable semi-annually in arrears on January 15 and July 15 of each year. The Notes matured and were paid in full on January 15, 2021.
10. Revenue recognition
The Company operates as one operating segment. Therefore, results of its operations are reported on a consolidated basis for purposes of segment reporting, consistent with internal management reporting.
The Company's revenues disaggregated by the major sources were as follows:
| Three Months Ended September 30, 2021 | Three Months Ended September 30, 2020 | ||||||||||||||||||||||||||||||||||
U.S. Government | Non-U.S. Government | Total | U.S. Government | Non-U.S. Government | Total | ||||||||||||||||||||||||||||||
| Product sales, net | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||||||||||
| Contract development and manufacturing: | |||||||||||||||||||||||||||||||||||
| Services | |||||||||||||||||||||||||||||||||||
| Leases | ( | ( | |||||||||||||||||||||||||||||||||
| Total contract development and manufacturing | ( | ||||||||||||||||||||||||||||||||||
| Contracts and grants | |||||||||||||||||||||||||||||||||||
| Total revenues | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||||||||||
| Nine Months Ended September 30, 2021 | Nine Months Ended September 30, 2020 | ||||||||||||||||||||||||||||||||||
U.S. Government | Non-U.S. Government | Total | U.S. Government | Non-U.S. Government | Total | ||||||||||||||||||||||||||||||
| Product sales, net | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||||||||||
| Contract development and manufacturing: | |||||||||||||||||||||||||||||||||||
| Services | |||||||||||||||||||||||||||||||||||
| Leases | |||||||||||||||||||||||||||||||||||
| Total contract development and manufacturing | |||||||||||||||||||||||||||||||||||
| Contracts and grants | |||||||||||||||||||||||||||||||||||
| Total revenues | $ | $ | $ | $ | $ | $ | |||||||||||||||||||||||||||||
During the three and nine months ended September 30, 2020, the Company entered into CDMO service arrangements with innovators in support of the COVID-19 pandemic resulting in an increase in non-government revenues during the three and nine months ended September 30, 2021 compared to the three and nine months ended September 30, 2020.
BARDA COVID-19 Development Public-Private Partnership
In 2020, we announced that we had been issued a task order under our existing Center for Innovation in Advanced Development and Manufacturing ("CIADM") agreement with BARDA for COVID-19 vaccine development and manufacturing (the "BARDA COVID-19 Development Public Private Partnership"). The initial task order had a contract value of up to $628.2 million and includes the reservation of manufacturing capacity and accelerated expansion of fill/finish capacity valued at $542.7 million and $85.5 million, respectively. Subsequently, the task order was expanded to include incremental capital activities which increased the value to $650.8 million. During the three months ended September 30, 2021, the Company has concluded that the BARDA COVID-19 Development Public Private Partnership
20
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
should be classified as a lease as BARDA received the right to direct the use of and obtain substantially all of the economic benefits of manufacturing suites (see Note 2). The lease arrangement with BARDA allows for the reservation of drug substance and drug product manufacturing capacity at various manufacturing sites that are recognized on a straight-line basis over the applicable lease term.
During the three months ended September 30, 2021, the Company determined that collection of all outstanding lease payments under our BARDA COVID-19 Development Public Private Partnership agreement was not probable, due to the passage of time as the USG had not paid invoices related to the task order reservation for services received after February 2021. The Company has reversed $86.0 million of revenue from prior periods during the three months ended September 30, 2021, which represents the amount of lease income recognized to date that is in excess of cash collected. Currently, the Company expects to record future lease payments on a cash basis. During the nine months ended September 30, 2021, the Company has recognized lease revenue of $81.9 million under this arrangement. During the three and nine months ended September 30, 2020, the Company recognized lease revenue of $85.9 million and $130.5 million, respectively under this arrangement.
On November 1, 2021, the Company and BARDA mutually agreed to terminate the Company's CIADM contract and associated task orders, including the BARDA COVID-19 Development Public Private Partnership. The terms of the contract modification reduced the contract value of the BARDA COVID-19 Development Public-Private Partnership to $470.9 million from $650.8 million and the base CIADM contract value was reduced to $140.5 million from $163.2 million. As a result of this termination, the Company expects to record approximately $215.9 million of revenue in the fourth quarter of 2021, which includes $155.7 million of lease revenue upon receipt of cash payments and $60.2 million of contract and grant revenue from the original CIADM arrangement, of which $55.2 million is recorded as deferred revenue as of September 30, 2020. In addition, the Company expects to recognize as research and development expense of $38.2 million due to the removal of a contract asset associated with the CIADM arrangement. Upon termination, the Company and BARDA have no ongoing obligations related to this arrangement.
Non-USG Leases
The Company's multi-year CDMO service arrangements with non-USG customers that were entered into during 2020 include operating leases whereby the customer has the right to direct the use of and obtain substantially all of the economic benefits of manufacturing suites. The associated revenue is recognized on a straight-line basis over the term of the lease. The remaining term on the Company's operating lease components approximates 2.5 years. The Company has allocated contracted operating lease revenues due under our long-term CDMO service arrangements with non-USG customers as follows:
21
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
| Year ended December 31, | |||||
2021 (1) | $ | ||||
| 2022 | |||||
| 2023 | |||||
| 2024 | |||||
| $ | |||||
(1) As of September 30, 2021, amount represents the three months ending December 31, 2021. | |||||
Transaction price allocated to remaining performance obligations
As of September 30, 2021, the Company expects future revenues on unsatisfied performance obligations of approximately $1.5 billion associated with all arrangements entered into by the Company. The unsatisfied performance obligations increased by $0.2 billion during the three months ended September 30, 2021, largely due to the receipt of a $0.4 billion contract modification to the 2016 AV7909 development and procurement contract with the U.S. Department of Health and Human Services ("HHS") offset by revenue recognized during the period.
During the three months ended June 30, 2021, AstraZeneca ("AZ") instructed the Company to cease performing new manufacturing services. At that time the Company updated its estimated transaction price subject to constraints and measure of progress under the arrangement. The Company has completed the release of all previously manufactured batches for AZ and there are no remaining unsatisfied performance obligations included in the Company's unsatisfied performance obligation disclosure.
The Company expects to recognize a majority of the $1.5 billion of unsatisfied performance obligations within the next 24 months. The amount and timing of revenue recognition for unsatisfied performance obligations can change. The future revenues associated with unsatisfied performance obligations exclude the value of unexercised option periods in the Company’s revenue arrangements. Often the timing of manufacturing activities changes based on customer needs and resource availability. Regulatory compliance may also impact the status of the Company’s COVID related CDMO arrangements. Government funding appropriations can impact the timing of product deliveries. The success of the Company's development activities that receive development funding support from the USG under development contracts can also impact the timing of revenue recognition.
Contract assets
The Company considers unbilled accounts receivables and deferred costs associated with revenue generating contracts, which are not included in inventory or property, plant and equipment, as contract assets. As of September 30, 2021 and December 31, 2020, the Company had contract assets associated with deferred costs of $38.0 million and $41.1 million, respectively, which is reflected as a component of other assets on the Company's consolidated balance sheets. During the three and nine months ended September 30, 2021, the Company recorded amortization expense of contract assets of $1.7 million and $3.3 million, respectively, which has been included as a component of research and development expense. The Company did no t record amortization expense associated with its contract assets during 2020.
Contract liabilities
When performance obligations are not transferred to a customer at the end of a reporting period, cash received associated with amounts allocated to those performance obligations is reflected as contract liabilities on the consolidated balance sheets and is deferred until control of these performance obligations is transferred to the customer. The following table presents the roll forward of the contract liability balances:
| December 31, 2020 | $ | ||||
| Deferral of revenue | |||||
| Revenue recognized | ( | ||||
| September 30, 2021 | $ | ||||
As of September 30, 2021 and December 31, 2020, the current portion of contract liabilities was $36.8 million and $44.6 million, respectively, and was included in other current liabilities on the balance sheet.
22
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
Accounts receivable
Accounts receivable, including unbilled accounts receivable contract assets, consist of the following:
| September 30, 2021 | December 31, 2020 | |||||||||||||
| Billed, net | $ | $ | ||||||||||||
| Unbilled | ||||||||||||||
| Total, net | $ | $ | ||||||||||||
11. Income taxes
12. Net (loss) income per share
The following table presents the calculation of basic and diluted net (loss) income per share:
| Three Months Ended September 30, | Nine Months Ended September 30, | |||||||||||||||||||
| 2021 | 2020 | 2021 | 2020 | |||||||||||||||||
| Numerator: | ||||||||||||||||||||
| Net (loss) income | $ | ( | $ | $ | $ | |||||||||||||||
| Denominator: | ||||||||||||||||||||
| Weighted-average number of shares—basic | ||||||||||||||||||||
| Dilutive securities—equity awards | ||||||||||||||||||||
| Weighted-average number of shares—diluted | ||||||||||||||||||||
| Net (loss) income per share - basic | $ | ( | $ | $ | $ | |||||||||||||||
| Net (loss) income per share - diluted | $ | ( | $ | $ | $ | |||||||||||||||
Basic net (loss) income per share is computed by dividing net (loss) income by the weighted average number of shares of common stock outstanding during the period. Diluted (loss) income per share is computed using the treasury method by dividing net (loss) income by the weighted average number of shares of common stock outstanding during the period, adjusted for the potential dilutive effect of other securities if such securities were converted or exercised and are not anti-dilutive. No adjustment for the potential dilutive effect of dilutive securities is reported for the three months ended September 30, 2021 as the effect would have been anti-dilutive due to the Company's net loss.
The following table presents the share-based awards that are not considered in the diluted net (loss) income per share calculation because the exercise price of the awards was greater than the average per share closing price during the three and nine months ended September 30, 2021 and 2020.
| Three Months Ended September 30, | Nine Months Ended September 30, | |||||||||||||||||||
| 2021 | 2020 | 2021 | 2020 | |||||||||||||||||
| Anti-dilutive stock awards | ||||||||||||||||||||
23
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
13. Equity
Share-based compensation
During the nine months ended September 30, 2021, the Company granted stock options to purchase 0.3 million shares of common stock and 0.6 million restricted and performance stock units under the Emergent BioSolutions Inc. Stock Incentive Plan. Typically, the stock option and restricted stock unit grants vest over three equal annual installments beginning on the day prior to the anniversary of the grant date. The performance stock units settle in stock at the end of the three-year performance period based on the Company's results compared to the performance criteria.
Share-based compensation expense was recorded in the following financial statement line items:
| Three Months Ended September 30, | Nine Months Ended September 30, | |||||||||||||||||||
| 2021 | 2020 | 2021 | 2020 | |||||||||||||||||
| Cost of product sales | $ | $ | $ | $ | ||||||||||||||||
| Cost of contract development and manufacturing | ||||||||||||||||||||
| Research and development | ||||||||||||||||||||
| Selling, general and administrative | ||||||||||||||||||||
| Total share-based compensation expense | $ | $ | $ | $ | ||||||||||||||||
Accumulated other comprehensive income (loss)
| (In Millions) | Defined Benefit Pension Plan | Derivative Instruments | Foreign Currency Translation Losses | Total | ||||||||||||||||||||||
Balance, December 31, 2020 | ( | ( | ( | ( | ||||||||||||||||||||||
| Other comprehensive (loss) income before reclassifications | — | ( | ( | ( | ||||||||||||||||||||||
| Amounts reclassified from accumulated other comprehensive income | — | — | ||||||||||||||||||||||||
Balance, September 30, 2021 | $ | ( | $ | ( | $ | ( | $ | ( | ||||||||||||||||||
Balance, June 30, 2021 | ( | ( | ( | ( | ||||||||||||||||||||||
| Other comprehensive (loss) income before reclassifications | — | ( | ( | |||||||||||||||||||||||
| Amounts reclassified from accumulated other comprehensive income | — | — | ||||||||||||||||||||||||
Balance, September 30, 2021 | $ | ( | $ | ( | $ | ( | $ | ( | ||||||||||||||||||
Balance, December 31, 2019 | ( | ( | ( | ( | ||||||||||||||||||||||
| Other comprehensive (loss) income before reclassifications | — | ( | ( | ( | ||||||||||||||||||||||
| Amounts reclassified from accumulated other comprehensive income | — | ( | — | ( | ||||||||||||||||||||||
Balance, September 30, 2020 | $ | ( | $ | ( | $ | ( | $ | ( | ||||||||||||||||||
Balance, June 30, 2020 | ( | ( | ( | ( | ||||||||||||||||||||||
| Other comprehensive (loss) income before reclassifications | — | ( | ||||||||||||||||||||||||
| Amounts reclassified from accumulated other comprehensive income | — | ( | — | ( | ||||||||||||||||||||||
Balance, September 30, 2020 | $ | ( | $ | ( | $ | ( | $ | ( | ||||||||||||||||||
During the three and nine months ended September 30, 2021, the tax impact related to unrealized gains (losses) on hedging activities was a (benefit) expense of $(2.2 ) million and $(1.4 ) million, respectively; the tax effects of the defined benefit pension plan and foreign currency translation losses were de minimis. During the three and nine ended September 30, 2020 there were tax (benefit) expense related to unrealized losses on hedging activities of
24
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
14. Commitments and contingencies
Securities and Shareholder Litigation
On April 20, 2021, May 14, 2021 and June 2, 2021 class action lawsuits were filed against the Company and certain of its current and former senior officers in the United States District Court for the District of Maryland on behalf of purchasers of the Company's common stock, seeking to pursue remedies under the Securities Exchange Act of 1934 (the “Exchange Act”). These complaints were filed by Plaintiff Palm Tran, Inc. – Amalgamated Transit Union Local 1577 Pension Plan; Plaintiff Alan I. Roth; and Plaintiff Stephen M. Weiss, respectively. The complaints allege, among other things, that the defendants disseminated materially false and misleading information about its capabilities to manufacture COVID-19 vaccine bulk drug substance in violation of Sections 10(b) and 20(a) of the Exchange Act and Rule 10b-5 promulgated thereunder. The defendants believe that the allegations in the complaints are without merit and intend to defend the matters vigorously.
It is expected that all three of these cases will be consolidated into a single action. Given the uncertainty of litigation, the preliminary stage of the cases, and the legal standards that must be met for, among other things, class certification and success on the merits, the Company cannot reasonably estimate the possible loss or range of loss, if any, that may result from these actions.
On June 29, 2021, Lincolnshire Police Pension Fund filed a stockholder derivative lawsuit in the United States District Court for the District of Maryland on behalf of the Company against certain of its current and former officers and directors for breach of fiduciary duties, waste of corporate assets, and unjust enrichment, each allegation related to the Company’s capabilities to manufacture COVID-19 vaccine bulk drug substance. In addition to monetary damages, the complaint seeks the implementation of multiple corporate governance and internal policy changes. The defendants believe that the allegations in the complaint are without merit and intend to defend the matter vigorously.
On September 15, 2021 and September 16, 2021, stockholder derivative lawsuits were filed by Chang Kyum Kim and Mark Nevins in The Court of Chancery of the State of Delaware on behalf of the Company against certain of its current and former officers and directors for breach of fiduciary duties, unjust enrichment and insider trading, each allegation related to the Company’s capabilities to manufacture COVID-19 vaccine bulk drug substance. In addition to monetary damages, the complaints seek the implementation of multiple corporate governance and internal policy changes. The defendants believe that the allegations in the complaints are without merit and intend to defend the matters vigorously. It is expected that both of these cases will be consolidated into a single action.
In addition to the above actions, the Company has received preliminary inquiries and subpoenas to produce documents related to these matters from Representative Maloney and Representative Clyburn, members of the Oversight Committee and the Select Subcommittee on the Coronavirus Crisis, Senator Murray of the Committee on Health, Education, Labor and Pensions, the Financial Industry Regulatory Authority (FINRA), the Department of Justice, the Securities and Exchange Commission (SEC), the Maryland Attorney General’s Office, and the New York Attorney General’s Office. The Company is producing and has produced documents as required in response and will continue to cooperate with these government inquiries.
Intellectual Property
Emergent BioSolutions’ Adapt Pharma subsidiaries (“Emergent”) are as follows: Emergent Devices Inc. (“EBPA”), formerly known as Adapt Pharma Inc.; Emergent Operations Ireland Limited (“EIRE”), formerly known as Adapt Pharma Operations Limited; and Emergent BioSolutions Ireland Limited (“EIR2”), formerly known as Adapt Pharma Limited.
ANDA Litigation - Teva 4mg
Emergent BioSolutions’ Adapt Pharma subsidiaries EBPA and EIRE, and Opiant Pharmaceuticals Inc. (“Opiant”) received notice letters from Teva Pharmaceuticals Industries Limited and Teva Pharmaceuticals USA (collectively, “Teva”) that Teva had filed an Abbreviated New Drug Application (“ANDA”) with the FDA seeking regulatory approval to market a generic version of NARCAN® (naloxone hydrochloride) Nasal Spray 4 mg/spray before the expiration of certain patents listed on the FDA’s website for Approved Drug Products with Therapeutic Equivalence Evaluations
25
EMERGENT BIOSOLUTIONS INC.
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, in millions, except share and per share amounts)
(“Orange Book Listed Patents”) for NARCAN®. Teva’s notice letters alleged that claims of certain Orange Book Listed Patents for NARCAN® were invalid and/or would not be infringed by the activities described in Teva’s ANDA. Emergent and Opiant filed complaints against Teva in the U.S. District Court for the District of New Jersey alleging infringement of certain Orange Book Listed Patents for NARCAN®. On June 5, 2020, the U.S. District Court for the District of New Jersey ruled in favor of Teva. Emergent appealed the District of New Jersey’s decision to the Court of Appeals for the Federal Circuit. The appeal hearing was held on August 2, 2021. A decision following the hearing is pending and anticipated before the end of 2021.
Emergent has also filed suit in the Federal Court in Canada against Teva Pharmaceuticals. The litigation in Canada is related to Teva Pharmaceuticals’ filing of an abbreviated new drug submission (“ANDS”) in Canada seeking to manufacture and sell a generic form of NARCAN® Nasal Spray ahead of the expiry of the Canadian patent covering our product. The trial date is currently scheduled for the end of March 2022.
ANDA Litigation - Teva 2mg
Emergent BioSolutions’ Adapt Pharma subsidiaries EBPA and EIRE, and Opiant received a notice letter from Teva that Teva had filed an ANDA with the FDA seeking regulatory approval to market a generic version of NARCAN® (naloxone hydrochloride) Nasal Spray 2 mg/spray before the expiration of certain Orange Book Listed Patents for the 2 mg/spray dose of NARCAN®. Teva’s notice letter alleged that claims of certain Orange Book Listed Patents for the 2 mg/spray dose of NARCAN® were invalid and/or would not be infringed by the activities described in Teva’s ANDA. Emergent and Opiant filed complaints against Teva in the U.S. District Court for the District of New Jersey alleging infringement of certain Orange Book Listed Patents for the 2 mg/spray dose of NARCAN®. This case is currently stayed pending the outcome of the appeal of the NARCAN® Nasal Spray 4 mg/spray case.
ITEM 2. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our unaudited condensed consolidated financial statements and accompanying notes and other financial information included elsewhere in this quarterly report on Form 10-Q and our audited consolidated financial statements and related notes included in our Annual Report on Form 10-K for the year ended December 31, 2020. Some of the information contained in this discussion and analysis or set forth elsewhere in this quarterly report on Form 10-Q, includes information with respect to our plans and strategy for our business and financing, as well as forward-looking statements that involve risks and uncertainties. You should carefully review the "Special Note Regarding Forward-Looking Statements" and "Risk Factors" sections of this quarterly report on Form 10-Q for a discussion of important factors that could cause actual results to differ materially from the results described in or implied by the forward-looking statements contained in the following discussion and analysis.
Business Overview
We are a global life sciences company focused on providing to civilian and military populations a portfolio of innovative preparedness and response products and solutions that address accidental, deliberate and naturally occurring PHTs.
We are currently focused on the following five distinct PHT categories: CBRNE, EID, travel health, emerging health crises, acute/emergency care; and CDMO. We have a product portfolio of ten products (vaccines, therapeutics, and drug-device combination products) that contribute a substantial portion of our revenue. We also have two procured product candidates that are procured under special circumstances by certain government agencies, although they are not approved by the FDA. Additionally, we have a development pipeline consisting of a diversified mix of both pre-clinical and clinical stage product candidates (vaccines, therapeutics, devices and combination products). Finally, we have a fully-integrated portfolio of contract development and manufacturing services. Our CDMO service offerings cover development services, drug substance manufacturing and drug product manufacturing across pharmaceutical and biotechnology industries as well as the USG and non-governmental organizations. The majority of our product revenue comes from the following products and procured product candidates:
26
EMERGENT BIOSOLUTIONS INC.
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
(unaudited, amounts in millions, except share and per share amounts)
Vaccines
•Anthrax vaccines, including our AV7909 (Anthrax Vaccine Adsorbed with Adjuvant) procured product candidate being developed as a next-generation anthrax vaccine for post-exposure prophylaxis and BioThrax® (Anthrax Vaccine Adsorbed), the only vaccine licensed by the FDA for the general use prophylaxis and post-exposure prophylaxis of anthrax disease. AV7909 has not been approved by the FDA, but is procured by certain authorized government buyers for their use;
•ACAM2000® (Smallpox (Vaccinia) Vaccine, Live), the only single-dose smallpox vaccine licensed by the FDA for active immunization against smallpox disease for persons determined to be at high risk for smallpox infection;
•Vivotif® (Typhoid Vaccine Live Oral Ty21a), the only oral vaccine licensed by the FDA for the prevention of typhoid fever; and
•Vaxchora® (Cholera Vaccine, Live, Oral), the only single-dose oral vaccine approved by the FDA and EMA for the prevention of cholera.
Devices
•NARCAN® (naloxone HCl) Nasal Spray, the first needle-free formulation of naloxone approved by the FDA and Health Canada, for the emergency treatment of known or suspected opioid overdose as manifested by respiratory and/or central nervous system depression;
•RSDL® (Reactive Skin Decontamination Lotion Kit), the only medical device cleared by the FDA to remove or neutralize the following chemical warfare agents from the skin: tabun, sarin, soman, cyclohexyl sarin, VR, VX, mustard gas and T-2 toxin; and
•Trobigard®, a combination drug-device auto-injector procured product candidate that contains atropine sulfate and obidoxime chloride. It has not been approved by the FDA, but is procured by certain authorized government buyers under special circumstances for potential use as a nerve agent countermeasure.
Therapeutics
•raxibacumab (Anthrax Monoclonal), the first fully human monoclonal antibody therapeutic licensed by the FDA for the treatment and prophylaxis of inhalational anthrax;
•Anthrasil® (Anthrax Immune Globulin Intravenous (Human)), the only polyclonal antibody therapeutic licensed by the FDA and Health Canada for the treatment of inhalational anthrax;
•BAT® (Botulism Antitoxin Heptavalent (A,B,C,D,E,F,G)-(Equine)), the only heptavalent antibody therapeutic licensed by the FDA and Health Canada for the treatment of botulism; and
•VIGIV (Vaccinia Immune Globulin Intravenous (Human)), the only polyclonal antibody therapeutic licensed by the FDA and Health Canada to address certain complications from smallpox vaccination.
Contract Development and Manufacturing
Our portfolio of CDMO revenues consists of distinct but interrelated service groups: development services (process and analytical development); drug substance manufacturing; drug product manufacturing (fill/finish) and packaging; and, when necessary, suite reservation obligations. These services, which we refer to as "molecule-to-market" offerings, employ five technology platforms (mammalian, microbial, viral, plasma and gene therapy) across a network of nine geographically distinct development and manufacturing sites operated by us for our internal products and pipeline and CDMO services, for both clinical-stage and commercial-stage projects. We direct these CDMO activities for a variety of third-party customers, including government agencies, innovative pharmaceutical companies, and non-government organizations.
Organizational Structure Changes
In October 2021, the Company implemented a new organizational structure organized around markets and customers whereas our historical structure was organized around product/platform and service types. The key components of the new business structure include a Government (MCM) line of products, Commercial line of products, and CDMO offerings as well as the centralization of R&D functions and capabilities at the enterprise level. In future filings, we will categorize and describe our business in accordance with the new structure. We will continue to provide similar information regarding our products and our financial statement presentation will remain the same. The Company will continue to have one reportable segment based on the management of the business and information reviewed by the Company’s Chief Operating Decision Maker ("CODM"), our President and Chief Executive Officer.
27
EMERGENT BIOSOLUTIONS INC.
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
(unaudited, amounts in millions, except share and per share amounts)
Financial Operations Overview
Revenues
We generate product revenues from the sale of our marketed products and procured product candidates which include vaccines, therapeutics and devices which have been described above. The USG is the largest purchaser of our CBRNE products and primarily purchases our products for the SNS, a national repository of medical countermeasures including critical antibiotics, vaccines, chemical antidotes, antitoxins, and other critical medical supplies. The USG primarily purchases our products under long-term, firm fixed-price procurement contracts, generally with annual options. Our opioid overdose reversal product, NARCAN® Nasal Spray and our travel health products, Vivotif and Vaxchora, are sold commercially through wholesalers and distributors, physician-directed or standing order prescriptions at retail pharmacies, and to state and local community healthcare agencies, practitioners and hospitals.
We also generate revenue from our CDMO services, which is based on our established development and manufacturing infrastructure, technology platforms and expertise. Our services include a fully integrated molecule-to-market contract development and manufacturing services business offering across development services, drug substance and drug product for small to large pharmaceutical and biotechnology industry and government agencies/non-governmental organizations. From time to time, clients require suite reservations at our various manufacturing sites, which may be considered leases depending on the facts and circumstances.
We have received contracts and grants funding from the USG and other non-governmental organizations to perform research and development activities, particularly related to programs addressing certain CBRNE threats and EIDs.
Our revenue, operating results and profitability vary quarterly based on the timing of production and deliveries and the nature of our business to provide large scale bundles of products and services as needs arise. Since early 2020, our revenues from the sale of our vaccine products that target travelers have also declined due to the reduction of international travel caused by the COVID-19 pandemic. We expect continued variability in our quarterly financial statements.
Cost of Sales
Products - The primary expenses that we incur to deliver our products consist of fixed and variable costs. We determine the cost of product sales for products sold during a reporting period based on the average manufacturing cost per unit in the period those units were manufactured. Fixed manufacturing costs include facilities, utilities and amortization of intangible assets. Variable manufacturing costs primarily consist of costs for materials and personnel-related expenses for direct and indirect manufacturing support staff, contract manufacturing operations, sales-based royalties, shipping and logistics. In addition to the fixed and variable manufacturing costs described above, the cost of product sales depends on utilization of available manufacturing capacity. For our commercial sales, other associated expenses include sales-based royalties (which include fair value adjustments associated with contingent consideration), shipping, and logistics.
CDMO - The primary expenses that we incur to deliver our CDMO services consist of fixed and variable costs. We operate five facilities that perform manufacturing activities for CDMO services customers. We use the same manufacturing facilities and methods of production for our own products as well as for fulfillment of our CDMO service contracts. Our manufacturing process includes the production of bulk material and performing “fill finish” work for containment and distribution of biological products. For “fill finish” customers, we receive work in process inventory to be prepared for distribution. When producing bulk material, we generally procure raw materials, manufacture the product and retain the risk of loss through the manufacturing and review process until delivery.
Research and Development Expenses
We expense research and development costs as incurred. Our research and development expenses consist primarily of:
▪personnel-related expenses;
▪fees to professional service providers for, among other things, analytical testing, independent monitoring or other administration of our clinical trials and obtaining and evaluating data from our clinical trials and non-clinical studies;
▪costs of CDMO services for clinical trial material; and
▪costs of materials used in clinical trials and research and development.
28
EMERGENT BIOSOLUTIONS INC.
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
(unaudited, amounts in millions, except share and per share amounts)
In many cases, we plan to seek funding for development activities from external sources and third parties, such as governments and non-governmental organizations, or through collaborative partnerships. We expect our research and development spending will be dependent upon such factors as the results from our clinical trials, the availability of reimbursement of research and development spending, the number of product candidates under development, the size, structure and duration of any clinical programs that we may initiate, the costs associated with manufacturing and development of our product candidates on a large-scale basis for later stage clinical trials, and our ability to use or rely on data generated by government agencies.
Selling, General and Administrative Expenses
Selling, general and administrative expenses consist primarily of personnel-related costs and professional fees in support of our executives, sales and marketing, business development, government affairs, finance, accounting, information technology, legal, human resource functions and other corporate functions. Other costs include facility costs not otherwise included in cost of product sales and CDMO services or research and development expense.
Income Taxes
Uncertainty in income taxes is accounted for using a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. We recognize in our financial statements the impact of a tax position if that position is more likely than not of being sustained on audit, based on the technical merits of the position.
Management believes that the assumptions and estimates related to the provision for income taxes are critical to the Company’s results of operations.
New Accounting Standards
For a discussion of new accounting standards please read Note 2. Basis of Presentation, to our condensed consolidated financial statements included in this report.
Critical Accounting Policies and Estimates
The preparation of our condensed consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the U.S., requires us to make estimates, judgments and assumptions that may affect the reported amounts of assets, liabilities, equity, revenues and expenses and related disclosure of contingent assets and liabilities. On an ongoing basis we evaluate our estimates, judgments and methodologies. We base our estimates on historical experience and on various other assumptions that we believe are reasonable, the results of which form the basis for making judgments about the carrying values of assets, liabilities and equity and the amount of revenues and expenses. Actual results may differ from these estimates. During the nine months ended September 30, 2021, there have been no significant changes to our critical accounting policies and estimates contained in our Annual Report on Form 10-K for the year ended December 31, 2020, as filed with the SEC, other than those outlined in Note 2 to the accompanying condensed consolidated financial statements.
29
EMERGENT BIOSOLUTIONS INC.
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION
(unaudited, amounts in millions, except share and per share amounts)
Results of Operations
| Three Months Ended September 30, | Nine Months Ended September 30, | ||||||||||||||||||||||||||||||||||||||||||||||
| 2021 | 2020 | $ Change | % Change | 2021 | 2020 | $ Change | % Change | ||||||||||||||||||||||||||||||||||||||||
| Product sales net: | |||||||||||||||||||||||||||||||||||||||||||||||
| NARCAN Nasal Spray | $ | 133.3 | $ | 88.8 | $ | 44.5 | 50 | % | $ | 313.7 | $ | 233.8 | $ | 79.9 | 34 | % | |||||||||||||||||||||||||||||||
| ACAM2000 | 80.7 | 1.0 | 79.7 | NM | 80.7 | 71.0 | 9.7 | 14 | % | ||||||||||||||||||||||||||||||||||||||
| Anthrax vaccines | 15.6 | 73.9 | (58.3) | (79) | % | 122.1 | 258.1 | (136.0) | (53) | % | |||||||||||||||||||||||||||||||||||||
| Other product sales | 40.9 | 38.5 | 2.4 | 6 | % | 73.1 | 86.0 | (12.9) | (15) | % | |||||||||||||||||||||||||||||||||||||
| Total product sales, net | 270.5 | 202.2 | 68.3 | 34 | % | 589.6 | 648.9 | (59.3) | (9) | % | |||||||||||||||||||||||||||||||||||||
| Contract development and manufacturing: | |||||||||||||||||||||||||||||||||||||||||||||||
| Services | 112.6 | 53.1 | 59.5 | NM | 283.7 | 102.7 | 181.0 | NM | |||||||||||||||||||||||||||||||||||||||
| Leases | (71.0) | 104.0 | (175.0) | NM | 132.6 | 148.7 | (16.1) | (11) | % | ||||||||||||||||||||||||||||||||||||||
| Total contract development and manufacturing | 41.6 | 157.1 | (115.5) | (74) | % | 416.3 | 251.4 | 164.9 | 66 | % | |||||||||||||||||||||||||||||||||||||
| Contracts and grants | 16.9 | 25.9 | (9.0) | (35) | % | 63.6 | 72.1 | (8.5) | (12) | % | |||||||||||||||||||||||||||||||||||||
| Total revenues | 329.0 | 385.2 | (56.2) | (15) | % | 1,069.5 | 972.4 | 97.1 | 10 | % | |||||||||||||||||||||||||||||||||||||
| Operating expenses: | |||||||||||||||||||||||||||||||||||||||||||||||
| Cost of product sales | 103.2 | 120.2 | (17.0) | (14 | %) | 237.0 | 287.6 | (50.6) | (18 | %) | |||||||||||||||||||||||||||||||||||||
| Cost of contract development and manufacturing | 114.3 | 28.8 | 85.5 | NM | 307.6 | 68.1 | 239.5 | NM | |||||||||||||||||||||||||||||||||||||||
| Research and development | 49.6 | 84.4 | (34.8) | (41 | %) | 151.0 | 175.0 | (24.0) | (14 | %) | |||||||||||||||||||||||||||||||||||||
| Selling, general and administrative | 82.1 | 75.5 | 6.6 | 9 | % | 254.2 | 221.2 | 33.0 | 15 | % | |||||||||||||||||||||||||||||||||||||
| Amortization of intangible assets | 14.5 | 15.0 | (0.5) | (3 | %) | 44.5 | 44.8 | (0.3) | (1 | %) | |||||||||||||||||||||||||||||||||||||
| Total operating expenses | 363.7 | 323.9 | 39.8 | 12 | % | 994.3 | 796.7 | 197.6 | 25 | % | |||||||||||||||||||||||||||||||||||||
| Income (loss) from operations | (34.7) | 61.3 | (96.0) | NM | 75.2 | 175.7 | (100.5) | (57 | %) | ||||||||||||||||||||||||||||||||||||||
| Other income (expense): | |||||||||||||||||||||||||||||||||||||||||||||||
| Interest expense | (8.4) | (7.6) | (0.8) | 11 | % | (25.5) | (22.6) | (2.9) | 13 | % | |||||||||||||||||||||||||||||||||||||
| Other, net | (2.4) | 1.3 | (3.7) | NM | (2.8) | 1.3 | (4.1) | NM | |||||||||||||||||||||||||||||||||||||||
| Total other income (expense), net | (10.8) | (6.3) | (4.5) | 71 | % | (28.3) | (21.3) | (7.0) | 33 | % | |||||||||||||||||||||||||||||||||||||
| Income (loss) before income taxes | (45.5) | 55.0 | (100.5) | NM | 46.9 | 154.4 | (107.5) | (70 | %) | ||||||||||||||||||||||||||||||||||||||
| Income taxes | 12.8 | (15.5) | 28.3 | NM | (5.3) | (34.7) | 29.4 | (85 | %) | ||||||||||||||||||||||||||||||||||||||
| Net income (loss) | $ | (32.7) | $ | 39.5 | $ | (72.2) | NM | $ | 41.6 | $ | 119.7 | $ | (78.1) | (65 | %) | ||||||||||||||||||||||||||||||||
| NM - Not meaningful | |||||||||||||||||||||||||||||||||||||||||||||||
30
EMERGENT BIOSOLUTIONS INC.
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION
(unaudited, amounts in millions, except share and per share amounts)
Total Revenues

| Legend | |||||||||||
| NARCAN nasal spray | CDMO services | ||||||||||
| ACAM2000 | CDMO Leases | ||||||||||
| Anthrax Vaccines | Contracts and Grants | ||||||||||
| Other product sales | |||||||||||
Product Sales, net
NARCAN Nasal Spray
The increase in NARCAN Nasal Spray sales for the three and nine months ended September 30, 2021 was driven by growth in unit sales to the U.S. public interest customers and to a lesser extent the commercial retail markets. Less sizable increases in Canadian sales due to increases in units sold also contributed to growth during these periods.
ACAM2000
The increase in ACAM2000 sales for the three and nine months ended September 30, 2021 was driven by a combination of the USG's exercise of its purchase option in July 2021 and the impact to the timing of deliveries to the USG. The price per unit of ACAM2000 was largely consistent period over period, and therefore the fluctuation in revenue is due to the
number of units delivered. ACAM2000 product sales are made under procurement contracts with the USG and fluctuation in revenues are dictated by the timing of delivery of orders.
Anthrax Vaccines
The decrease in Anthrax vaccine sales for the three and nine months ended September 30, 2021 was primarily due to the timing of deliveries to the U.S government. The price per unit of AV7909 was largely consistent period over period, and therefore the fluctuation in revenue is due to the number of units delivered. Anthrax vaccine product sales are made under annual purchase options exercised by the USG. Fluctuations in revenues result from the timing of the exercise of annual purchase options and the USG purchases and Company delivery of orders that follow. The USG modified its contract to purchase additional doses of AV7909 on September 30, 2021.
Other Product Sales
Other product sales for the three months ended September 30, 2021 remained consistent compared to the three months ended September 30, 2020, as increased sales of VIGIV were offset by decreased sales of BAT. During the nine months ended September 30, 2021 other product sales decreased primarily due to a decline in sales of BAT due to timing of deliveries to the SNS, partially offset by increases in various other products.
CDMO
Services
The increase in CDMO services revenue for the three months ended September 30, 2021 of $59.5 million is largely due to out-of-period adjustments (see Note 2) of $49.3 million along with incremental manufacturing activities. The increase for the nine months ended September 30, 2021 is due to the Company's partnerships with innovators, including JNJ and AZ, to address the COVID-19 pandemic. The AZ and JNJ arrangements were entered into during the second and third quarters of 2020 and the Company began servicing other innovator companies in 2021. Additionally, during the nine months ended September 30, 2021, there were out-of-period adjustments (see Note 2) of $28.8 million.
Leases
The decrease in CDMO lease revenue during the three months ended September 30, 2021 was primarily due to a $171.9 million reduction in lease revenue associated with the COVID-19 development public-private partnership with BARDA as the Company recognized revenue of $85.9 million in Q3 2020 and
31
EMERGENT BIOSOLUTIONS INC.
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION
(unaudited, amounts in millions, except share and per share amounts)
recorded a reversal of revenue of $86.0 million during Q3 2021 based on a collectibility assessment as of September 30, 2021. The Company and BARDA mutually terminated this arrangement in November 2021 and the Company will record lease revenue in the fourth quarter 2021 to reflect the terminated contract value.
The decrease in CDMO lease revenue during the nine months ended September 30, 2021 was primarily due to a decline in lease revenue associated with the COVID-19 development public-private partnership with BARDA of $48.7 million offset by an increase in lease revenues associated with the Company's arrangement with JNJ.
Contracts and Grants
Contracts and grants revenue for the three and nine months ended September 30, 2021 decreased as compared to the three and nine months ended September 30, 2020 largely due a decrease in activities associated with the Company's COVID-HIG therapeutic product candidate. The nine months ended September 30, 2021 were also impacted by decreases in developmental activities associated with AV7909.
Cost of Product Sales
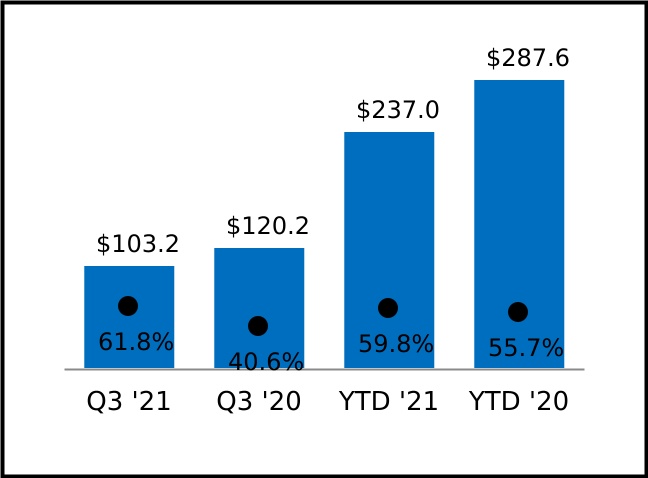
| Cost of product sales | |||||
| l | Gross profit margin for product sales | ||||
Cost of product sales decreased for the three and nine months ended September 30, 2021, due to contingent consideration charges for business combinations and inventory write-offs associated with the Company's travel health vaccines during the three and nine months ended September 30, 2020 that did not recur during the three and nine months ended September 30, 2021. This decrease was offset by higher volume of product sales, specifically NARCAN®
Nasal Spray and ACAM2000 during the three months ended September 30, 2021.
The increase in product margin percentage for the three and nine months ended September 30, 2021 is largely due to the impact of the non-recurring charges mentioned above. The gross margin was consistent during the three months ended September 30, 2021 excluding the non-recurring charges and decreased during the nine months ended September 30, 2021. The nine months ended September 30, 2021 was negatively impacted by product revenue mix which was weighted more heavily to lower margin products.
Cost of CDMO
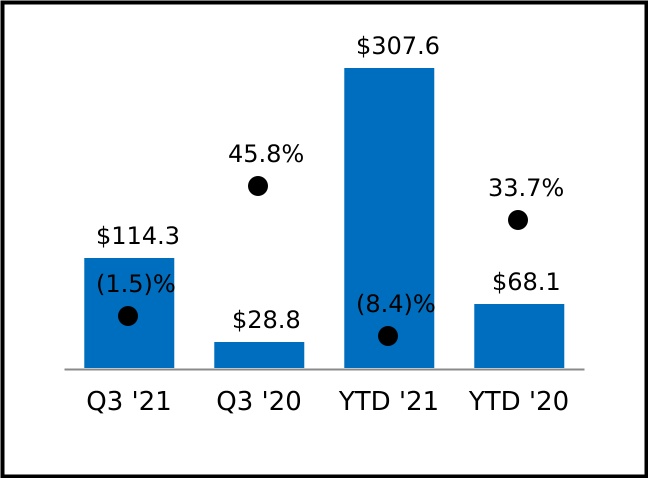
| Cost of CDMO services | |||||
| l | Gross profit margin for CDMO services | ||||
Cost of CDMO increased for the three and nine months ended September 30, 2021 as compared to the three and nine months ended September 30, 2020 due to an increase in CDMO service activities, increases in costs due to out-of-period adjustments (see Note 2) and additional costs to support remediation efforts for our COVID-19 manufacturing activities. Additionally, during the nine months ended September 30, 2021, the Company recorded inventory write-offs at its Bayview facility of $41.5 million, which were directly or indirectly the result of the cross-contamination event at the Bayview facility identified during the three months ended June 30, 2021.
32
EMERGENT BIOSOLUTIONS INC.
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION
(unaudited, amounts in millions, except share and per share amounts)
Research and Development Expenses (Gross and Net)

| Research and development expense | |||||
| l | Research and development expense, net of contracts and grants revenue | ||||
Research and development expense for the three and nine months ended September 30, 2021 decreased due to an impairment charge of $29.0 million related to the Company's IPR&D intangible asset recorded during the three months ended September 30, 2020. Additionally during the three months ended September 30, 2021 there was a decline in spending for the Company's COVID-HIG therapeutic product candidate and during the nine months ended September 30, 2021 there was decline in developmental activities associated with the Company's AV7909 product candidate.
Selling, General and Administrative Expenses
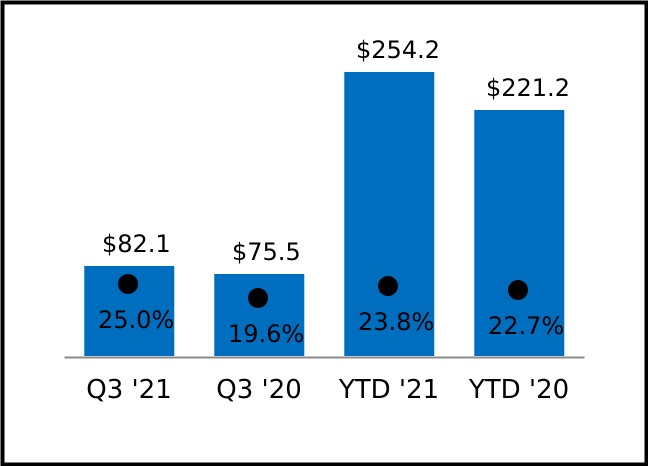
| Selling, general and administrative expenses | |||||
| l | SG&A as a percentage of total revenue | ||||
The increase in selling, general and administrative expenses for the three and nine months ended September 30, 2021 is due to an increase in headcount and professional services. The increase for the nine months ended September 30, 2021 is also impacted by increased costs for defending and supporting the Company's corporate reputation.
Amortization of Intangible Assets
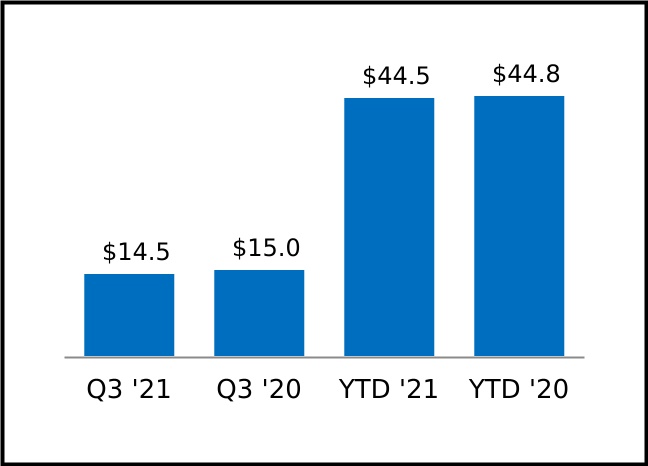
| Amortization expense | |||||
Amortization of intangible assets and the composition of intangible assets amortized during the three and nine months ended September 30, 2021 were consistent with the three and nine months ended September 30, 2020.
Other Income (Expense), Net
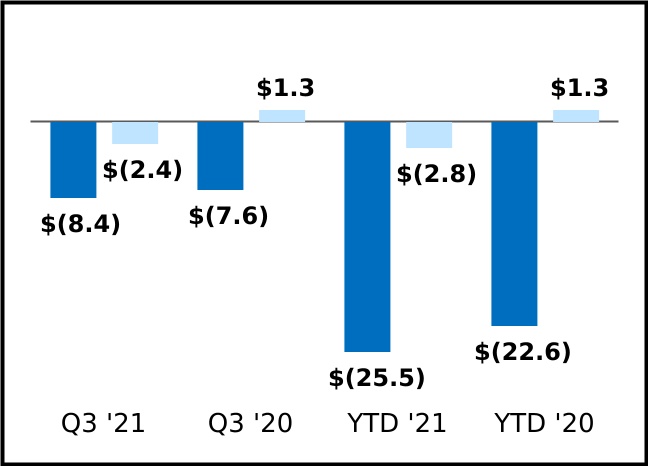
| Interest expense | |||||
| Other income (expense) | |||||
Total other income (expense), net decreased for the three and nine months ended September 30, 2021 largely due to increases in interest expense as a result
33
EMERGENT BIOSOLUTIONS INC.
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION
(unaudited, amounts in millions, except share and per share amounts)
of increases in total outstanding debt and interest rates during the periods.
Income Taxes
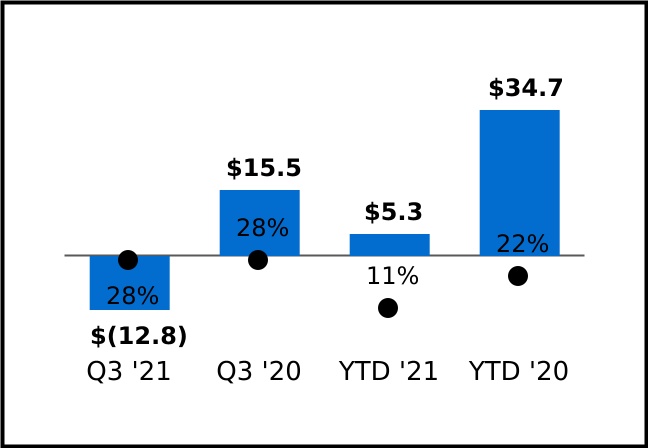
| Income taxes | |||||
| l | Effective tax rate | ||||
During the three and nine months ended September 30, 2021 and 2020, the estimated effective annual tax rates were 24% and 29%, respectively. The actual effective tax rates includes the impact of discrete tax benefits during the nine months ended September 30, 2021 and 2020 of $7.2 million and $9.9 million, respectively. Income taxes decreased during the periods largely due to the decline in income before income taxes.
Financial Condition, Liquidity and Capital Resources
Our financial condition is summarized as follows:
| (in millions, except percentages) | September 30, 2021 | December 31, 2020 | Change % | ||||||||||||||
| Financial assets: | |||||||||||||||||
| Cash and cash equivalents | $ | 403.8 | $ | 621.3 | (35) | % | |||||||||||
| Borrowings: | |||||||||||||||||
| Debt, current portion | 31.6 | 33.8 | (7) | % | |||||||||||||
| Debt, net of current portion | 817.3 | 841.0 | (3) | % | |||||||||||||
| Total borrowings | 848.9 | 874.8 | (3) | % | |||||||||||||
| Working capital: | |||||||||||||||||
| Current assets | 1,111.5 | 1,195.9 | (7) | % | |||||||||||||
| Current liabilities | 365.4 | 384.5 | (5) | % | |||||||||||||
| Total working capital | 746.1 | $ | 811.4 | (8) | % | ||||||||||||
Sources of Liquidity
We have historically financed our operating and capital expenditures through cash on hand, cash from operations, debt financing and contracts and grants development funding. We also obtain financing from the sale of our common stock upon exercise of stock options. We have operated profitably for each of the last five annual fiscal years through the period ended December 31, 2020. As of September 30, 2021, we had unrestricted cash and cash equivalents of
34
EMERGENT BIOSOLUTIONS INC.
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION
(unaudited, amounts in millions, except share and per share amounts)
$403.8 million and capacity under our revolving credit facility of $597.6 million. As of September 30, 2021, we believe that we have sufficient liquidity to fund our operations over the next 12 months.
Cash Flows
The following table provides information regarding our cash flows for the nine months ended September 30, 2021 and 2020:
| Nine Months Ended September 30, | |||||||||||
| 2021 | 2020 | ||||||||||
| Net cash provided by (used in): | |||||||||||
| Operating activities | $ | (7.9) | $ | 290.9 | |||||||
| Investing activities | (178.3) | (115.0) | |||||||||
| Financing activities | (31.0) | 71.8 | |||||||||
| Effect of exchange rate changes on cash, cash equivalents and restricted cash | (0.3) | (0.5) | |||||||||
| Net change in cash, cash equivalents and restricted cash | $ | (217.5) | $ | 247.2 | |||||||
Operating Activities
Net cash used in operating activities of $7.9 million for the nine months ended September 30, 2021 was due to net income excluding non-cash items of $266.0 million offset by negative working capital changes of $273.9 million due to increases in receivables and the accumulation of inventory and prepaid expenses.
Net cash provided by operating activities of $290.9 million for the nine months ended September 30, 2020 was due to net income excluding non-cash items of $305.2 million offset by working capital changes of $14.3 million.
The decrease of $298.8 in from cash provided by operating activities of $290.9 million to cash used in operating activities of $7.9 million is due to a decline in net income excluding non-cash items of $39.2 million and negative impacts from working capital changes of $259.6 million largely a result of changes in accounts receivables.
Investing Activities
Net cash used in investing activities relates to purchases of property, plant and equipment and was $178.3 million and $115.0 million for the nine months ended September 30, 2021 and 2020, respectively. The cash used in investing activities increased during the nine months ended September 30, 2021 largely due to infrastructure and equipment investments related to continued investments
associated with increased capacity and capabilities at our Rockville and Bayview facilities.
Financing Activities
Net cash used in financing activities of $31.0 million for the nine months ended September 30, 2021 was primarily due to payments on debt of $27.5 million.
Net cash provided by financing activities of $71.8 million for the nine months ended September 30, 2020 was primarily due to proceeds from the $450.0 million Senior Unsecured Notes offset by payments of $381.4 million on the term loan and revolving credit facility and $8.4 million of debt issuance costs.
The decrease of $102.8 million from cash provided by financing activities of $71.8 million to cash used in financing activities of $31.0 million is largely due to an increase of outflows due to debt activities during the nine months ended September 30, 2021 of $87.7 million.
Funding Requirements
We expect to continue to fund our anticipated operating expenses, capital expenditures, debt service requirements and any future repurchase of our common stock from the following sources:
▪existing cash and cash equivalents;
▪net proceeds from the sale of our products and contract development and manufacturing services;
▪development contracts and grants funding; and
35
EMERGENT BIOSOLUTIONS INC.
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATION
(unaudited, amounts in millions, except share and per share amounts)
▪our Senior Secured Credit Facilities and any other lines of credit we may establish from time to time.
There are numerous risks and uncertainties associated with product sales, delivery of CDMO services and with the development and commercialization of our product candidates. We may seek additional external financing to provide additional financial flexibility. Our future capital requirements will depend on many factors, including (but not limited to):
▪the level, timing and cost of product sales and cost of contract development and manufacturing services;
▪the extent to which we acquire or invest in and integrate companies, businesses, products or technologies;
▪the acquisition of new facilities and capital improvements to new or existing facilities;
▪the payment obligations under our indebtedness;
▪the scope, progress, results and costs of our development activities;
▪our ability to obtain funding from collaborative partners, government entities and non-governmental organizations for our development programs;
▪the extent to which we adopt a share repurchase program and repurchase shares of our common stock and;
▪the costs of commercialization activities, including product marketing, sales and distribution.
If our capital resources are insufficient to meet our future capital requirements, we will need to finance our cash needs through public or private equity or debt offerings, bank loans or collaboration and licensing arrangements.
If we raise funds by issuing equity securities, our stockholders may experience dilution. Public or bank debt financing, if available, may involve agreements that include covenants, like those contained in our Senior Unsecured Notes due 2028 and the Senior Secured Credit Facilities, which could limit or restrict our ability to take specific actions, such as incurring additional debt, making capital expenditures, pursuing acquisition opportunities, buying back shares or declaring dividends. If we raise funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish valuable rights to our technologies or product candidates or grant licenses on terms that may not be favorable to us.
Economic conditions, including market volatility and adverse impacts on financial markets as a result of the COVID-19 pandemic, may make it more difficult
to obtain financing on attractive terms, or at all. If financing is unavailable or lost, our business, operating results, financial condition and cash flows would be adversely affected, and we could be forced to delay, reduce the scope of or eliminate many of our planned activities.
Unused Credit Capacity
Available room under the revolving credit facility for the periods ended September 30, 2021 and December 31, 2020 was:
(in millions) | |||||||||||
Total Capacity | Outstanding Letters of Credit | Outstanding Indebtedness on Revolving Credit Facility | Unused Capacity | ||||||||
| September 30, 2021 | |||||||||||
| $600.0 | 2.4 | — | $597.6 | ||||||||
December 31, 2020 | |||||||||||
| $600.0 | 2.8 | — | $597.2 | ||||||||
36
EMERGENT BIOSOLUTIONS INC.
ITEM 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
For a discussion of additional risks arising from our operations, see “Item 1A-Risk Factors” in this quarterly report.
Market Risk
We have interest rate and foreign currency market risk. Because of the short-term maturities of our cash and cash equivalents, we believe that an increase in market rates would likely not have a significant impact on the realized value of our investments.
Interest Rate Risk
We have debt with a mix of fixed and variable rates of interest. Floating rate debt carries interest based generally on the eurocurrency rate, as defined in our Amended Credit Agreement, plus an applicable margin. We manage the impact of interest rate changes on our variable debt through derivative interest rate swap arrangements. Increases in interest rates could result in an increase in interest payments for debt that we have not hedged through our interest rate swap arrangements. See Note 9, "Debt," to the Notes of our condensed consolidated financial statements included in this 2021 Quarterly Report under the caption Item 1, "Financial Statements.”
We have assessed our exposure to changes in interest rates by analyzing the sensitivity to our operating results assuming various changes in market interest rates. A hypothetical increase of one percentage point in the eurocurrency rate as of September 30, 2021 would increase our interest expense by approximately $0.6 million annually.
Foreign Currency Exchange Rate Risk
We have exposure to foreign currency exchange rate fluctuations worldwide and primarily with respect to the Euro, Canadian dollar, Swiss franc and British pound. We manage our foreign currency exchange rate risk primarily by either entering into foreign currency hedging transactions or incurring operating expenses in the local currency in the countries in which we operate, to the extent practical. We currently do not hedge all of our foreign currency exchange exposure and the movement of foreign currency exchange rates could have an adverse or positive impact on our results of operations.
ITEM 4. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our chief executive officer and chief financial officer, evaluated the effectiveness of our disclosure controls and procedures as of September 30, 2021. The term "disclosure controls and procedures," as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934 (the Exchange Act), means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized, and reported, within the time periods specified in the SEC's rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the company's management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Based on the evaluation of our disclosure controls and procedures as of September 30, 2021, our chief executive officer and chief financial officer concluded that, as of such date, our disclosure controls and procedures were not effective, because we had a material weakness in our internal control over financial reporting related to the technical accounting assessment of specific attributes within complex revenue arrangements with our customers (“Revenue Accounting Issue”). A material weakness (as defined in Rule 12b-2 under the Exchange Act) is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement in our annual or interim financial statements will not be prevented or detected on a timely basis. While the Revenue Accounting Issue did not result in a material misstatement to our consolidated financial statements for any prior periods through and including September 30, 2021, there was a reasonable possibility that a material misstatement of our interim or annual financial statements would not be prevented or detected on a timely basis.
More specifically, the Revenue Accounting Issue relates to our technical accounting assessment of the BARDA COVID-19 Development Public Private
37
EMERGENT BIOSOLUTIONS INC.
Partnership and CDMO revenue contracts and related accounting judgments primarily focused on (a) the scoping of lease and non-lease components and (b) the recognition of revenue. While these accounting judgments were considered and documented during our initial analyses, we subsequently identified certain adjustments that were indicative of a control deficiency that we believe represents a material weakness in our internal control over financial reporting. Immaterial out-of-period adjustments have been made to the Company’s financial statements for the nine months ended September 30, 2021 as disclosed in Note 2 to the condensed consolidated financial statements.
We have initiated and begun to implement measures designed to improve our internal control over financial reporting related to accounting for complex revenue transactions, including additional training related to ASC 606 and ASC 842 technical accounting and identification of additional resources to support our assessments, where deemed appropriate and enhancing the degree of review, consultation and approval related to significant complex revenue transactions with a focus on the accounting judgments. In addition, we plan to apply this enhanced analysis to any new arrangements entered into prior to the end of the year ending December 31, 2021. As a result of these efforts and given that the deficiencies relate to specific adjustments that were made during the period ended September 30, 2021, we believe that the Revenue Accounting Issue may be remediated during the fourth quarter of 2021.
Changes in Internal Control Over Financial Reporting
There have been no changes in our internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act that occurred during the quarter ended September 30, 2021 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting, except for changes related to the Revenue Accounting Issue described above. The Company has implemented new controls and procedures to account for CDMO revenue on a cost to complete basis and to engage additional resources to support assessments for complex transactions.
PART II. OTHER INFORMATION
ITEM 1. LEGAL PROCEEDINGS
See "Item 1 of Part I, “Financial Statements — Notes to condensed consolidated financial statements — Note 14 — Commitments and contingencies.”
ITEM 1A. RISK FACTORS
The following risk factors and other information included in this Quarterly Report on Form 10-Q should
be carefully considered. The occurrence of any of the following risks or of unknown risks and uncertainties may adversely affect our business, operating results and financial condition.
RISK FACTOR SUMMARY
There are a number of government contracting risks that could impact our business, financial condition, operating results and cash flows, including:
•Reduced demand for and/or funding for procurement of AV7909 and/or BioThrax or ACAM2000 and discontinuation of funding of our other USG procurement and development contracts.
•Failure to receive FDA licensure of AV7909 in a timely manner or at all.
•Failure to comply with laws and regulations pertaining to government contracts and resources required for responding to related government inquiries.
There are a number of product development and commercialization risks that could impact our business, financial condition, operating results and cash flows, including:
•Our inability to maintain quality and manufacturing compliance at our manufacturing facilities could hinder our ability to continue manufacturing COVID-19-related product candidates for our CDMO customers.
•The COVID-19 product candidates we are working on may not be safe or effective and we may be unable to manufacture sufficient quantities to meet demand.
•Clinical trials of product candidates are expensive and time-consuming, and their outcome is uncertain.
•We may fail to capitalize on the most scientifically, clinically or commercially promising or profitable product candidates.
Due to numerous factors, the COVID-19 coronavirus pandemic could have a material adverse impact on our business, results of operations and financial performance, including:
•Changes in government priorities resulting from the pandemic, supply chain shortages and changing employee working arrangements could impact our overall business.
•The evolving nature of COVID-19 and related vaccines and treatments and resulting changes in demand for such product candidates may impact sales of related services offered by our CDMO business.
38
EMERGENT BIOSOLUTIONS INC.
There are a number of regulatory and compliance risks that could impact our business, financial condition, operating results and cash flows, including:
•Conditions associated with approvals and ongoing regulation of products may limit how and to the extent we manufacture and market them.
•Failure to comply with various health care laws could result in substantial penalties.
•Failure to comply with obligations under U.S. governmental pricing programs may require reimbursement for underpayments and the payment of substantial penalties, sanctions and fines.
•The authority to sell unapproved MCMs to certain government entities can be ambiguous and subject us to regulatory enforcement actions.
There are a number of manufacturing risks that could impact our business, financial condition, operating results and cash flows, including:
•Disruption at, damage to or destruction of our development and/or manufacturing facilities may impede our ability to manufacture our products, as well as deliver our CDMO services.
•Our operations, including our use of hazardous materials, chemicals, bacteria and viruses expose us to significant potential liabilities.
There are a number of competitive and political risks that could impact our business, financial condition, operating results and cash flows, including:
•Development and commercialization of pharmaceutical products are subject to evolving private and public sector competition.
•NARCAN® Nasal Spray may be subject to additional branded and new generic competition.
•Biologic Products may be affected by the approval and entry of follow-on biologics, or biosimilars in the United States and other jurisdictions.
There are a number of risks related to our intellectual property that could impact our business, financial condition, operating results and cash flows, including:
•Challenges in defense or enforcement of our intellectual property rights, including against current or potential infringers.
• Potential discrepancies or challenges with respect to third party licenses.
• Potential loss of proprietary information and know-how, which carries the risk of reducing the value of our technology and products.
• Entry of competing generic drugs upon patent expiry or with patents no longer in force.
There are a number of risks related to reliance on third parties that could impact our business, financial condition, operating results and cash flows, including:
•The loss of sole-source suppliers or an increase in the price of inventory.
•If third parties do not perform as contractually required or as expected, we may not be able to obtain regulatory approval for or commercialize our product candidates.
There are a number of legal and reputational risks that could impact our business, financial condition, operating results and cash flows, including:
•Pending litigation and legal proceedings and the impact of any finding of liability or damages could adversely impact our business, financial condition and results of operations.
•Our work on public health threats has exposed us to criticism and may expose us to further criticism, from the media, government personnel, and others, which could further harm our reputation, negatively effect on our share price, operations, and our ability to attract and retain talent.
•The potential for cyber security incidents to harm our ability to operate our business effectively in light of our heightened risk profile.
•Inherent product liability exposure due to our unique business.
There are a number of financial risks that could impact our business, financial condition, operating results and cash flows, including:
•Our ability to maintain sufficient cash flow from our operations to pay our substantial debt, both now and in the future.
•Our ability to obtain additional funding and be able to raise capital when needed.
•Our ability to comply with the covenants under our Senior Secured Credit Facilities and other debt agreements.
There are a number of risks related to our strategic acquisitions and collaborations that could impact our business financial condition, operating results and cash flows, including:
39
EMERGENT BIOSOLUTIONS INC.
•Our strategy of generating growth through acquisitions may be unsuccessful.
•Our failure to successfully integrate acquired businesses and/or assets into our operations and our ability to realize the benefits of such acquisitions.
There are a number of risks associated with our common stock, including, but not limited to:
•Our business or our share price could be negatively affected as a result of the actions of shareholders.
•Due to his substantial ownership percentage, our Executive Chairman has the ability to exert significant influence over us with respect to the election of the members of our Board of Directors and to delay or prevent a change of control of us.
•The price of our common stock has been and remains subject to extreme volatility.
The risk factors below contain more detailed descriptions of the risks identified above, which may materially harm our business, financial condition or results of cash flows.
GOVERNMENT CONTRACTING RISKS
We currently derive a substantial portion of our revenue from USG procurement of AV7909 and ACAM2000 and have historically derived a substantial portion of our revenue from USG procurement of BioThrax. If the USG’s demand for and/or funding for procurement of AV7909 and/or BioThrax or ACAM2000 is substantially reduced, our business, financial condition, operating results and cash flows would be materially harmed.
We derive a substantial portion of our current and expected future revenues from USG procurement of AV7909. As AV7909 is a product development candidate, there is a higher level of risk that we may encounter challenges causing delays or an inability to deliver AV7909 than with BioThrax, which may have a material effect on our ability to generate and recognize revenue.
The success of our business and our future operating results are significantly dependent on anticipated funding for the procurement of our anthrax vaccines and the terms of such procurement by the USG, including the price per dose, the number of doses and the timing of deliveries. We have no certainty that funding will be made available for the procurement of our anthrax vaccines. If priorities for the SNS change generally, or as a result of the conclusion of the USG’s recently announced audit of the SNS, or with respect to the level of procurement of
our anthrax vaccines, funding to procure future doses of AV7909 or BioThrax may be delayed, limited or not available, BARDA may never complete the anticipated full transition to stockpiling AV7909 in support of anthrax preparedness, and our future business, financial condition, operating results and cash flows could be materially harmed.
In addition, we currently derive a substantial portion of our revenues from sales of ACAM2000 to the USG. If priorities for the SNS change with respect to ACAM2000 or the USG decides not to exercise additional options under our ACAM2000 contract, our future business, financial condition, operating results and cash flows could be materially harmed.
We may not receive eventual FDA licensure of AV7909 in a timely manner or at all. Delays in our ability to achieve a favorable outcome from the FDA could prevent us from realizing the full potential value of our BARDA contract for the advanced development and procurement of AV7909.
In collaboration with us, the CDC filed with the FDA a pre-Emergency Use Authorization (EUA) submission package related to AV7909, which enables FDA review of data in anticipation of a request for an EUA. This submission triggered BARDA to exercise its first contract option in July 2019 to procure 10 million doses of AV7909, its second contract option in July 2020 and, most recently, to fund another procurement commitment in October 2021 for inclusion of additional doses into the SNS in support of anthrax preparedness.
We are also working on a BLA for filing with the FDA related to AV7909 and plan to commence our submission of the BLA to the FDA by the end of this year. There can be no guarantee that we will meet our target date for submission. Moreover, even if we do, the FDA may decide that our data are insufficient and require additional pre-clinical, clinical or other studies. If we are unsuccessful in obtaining FDA licensure, in a timely manner or at all, we may not be able to realize the full potential value of the contract, which could have a material adverse effect on our future business, financial condition, operating results and cash flows.
Our USG procurement and development contracts require ongoing funding decisions by the USG. Simultaneous reduction or discontinuation of funding of these contracts could cause our business, financial condition, operating results and cash flows to suffer materially.
The USG is the principal customer for our PHT-focused MCMs and the primary source of funds for the development of most of our product candidates in our development pipeline, most notably our AV7909 procured product candidate. We anticipate that the USG will also be a principal customer for those MCMs
40
EMERGENT BIOSOLUTIONS INC.
that we successfully develop within our existing product development pipeline, as well as those we acquire in the future. Additionally, a significant portion of our revenue comes from USG development contracts and grants. Over its lifetime, a USG procurement or development program may be implemented through the award of many different individual contracts and subcontracts. The funding for such government programs is subject to Congressional appropriations, generally made on a fiscal year basis, even for programs designed to continue for several years. For example, procurement of AV7909 to be supplied under our development and procurement contract with BARDA are subject to the availability of funding, mostly from annual appropriations. These appropriations can be subject to political considerations, changes in priorities due to global pandemics, the results of elections and stringent budgetary constraints.
Additionally, our government-funded development contracts typically give the USG the right, exercisable in its sole discretion, to extend these contracts for successive option periods following a base period of performance. The value of the services to be performed during these option periods may constitute the majority of the total value of the underlying contract. For example, the September 2016 contract award from BARDA for the development and delivery to the SNS of AV7909 for post-exposure prophylaxis of anthrax disease consists of a five-year base period of performance. The contract award also includes options for the delivery of additional doses of AV7909 to the SNS and options for an additional clinical study and post-marketing commitments. This contract was recently extended through 2025, and provides for additional procurement of AV7909 for the SNS over the next 18 months. If levels of government expenditures and authorizations for public health countermeasure preparedness decrease or shift to programs in areas where we do not offer products or are not developing product candidates, or if the USG otherwise declines to exercise its options under our existing contracts, our revenues would suffer, as well as our business, financial condition, operating results and cash flows.
There can be no assurance that we will be able to secure follow-on procurement contracts with the USG upon the expiration of any of our product procurement contracts.
A significant portion of our revenue is substantially dependent upon product procurement contracts with the USG and foreign governments for our PHT products. Upon the expiration of a procurement contract, we may not be able to negotiate a follow-on procurement contract for the particular product for a similar product volume, period of performance, pricing or other terms, or at all. The inability to secure a similar or increased procurement contract could materially affect our
revenues and our business, financial condition, operating results and cash flows could be harmed. For example, in November 2019, the BARDA procurement contract for raxibacumab that we acquired in our 2017 acquisition of the product from GlaxoSmithKline LLC was completed. We intend to negotiate a follow-on procurement contract for raxibacumab and other follow-on procurement contracts for most of our PHT products upon the expiration of a related procurement contract, but there can be no assurance that we will be successful obtaining any follow-on contracts. Even if we are successful in negotiating a follow-on procurement contract, it may be for a lower product volume, over a shorter period of performance or be on less favorable pricing or other terms. An inability to secure follow-on procurement contracts for our products or procured product candidates could materially and adversely affect our revenues, and our business, financial condition, operating results and cash flows could be harmed.
The government contracting process is typically a competitive bidding process and involves unique risks and requirements.
Our business involves government contracts and grants, which may be awarded through competitive bidding. Competitive bidding for government contracts presents many risks and requirements, including:
•the possibility that we may be ineligible to respond to a request for proposal;
•the commitment of substantial time and attention of management and key employees to the preparation of bids and proposals;
•the need to accurately estimate the resources and cost structure that will be required to perform any contract that we might be awarded;
•the submission by third parties of protests to our responses to requests for proposal that could result in delays or withdrawals of those requests for proposal; and
•in the event our competitors protest or challenge contract or grant awards made to us through competitive bidding, the potential that we may incur expenses or delays, and that any such protest or challenge could result in the resubmission of bids based on modified specifications, or in the termination, reduction or modification of the awarded contract.
The USG may choose not to award us future contracts for either the development of our new product candidates or for the procurement of our existing products addressing PHTs and may instead award such contracts to our competitors. If we are
41
EMERGENT BIOSOLUTIONS INC.
unable to secure particular contracts, we may not be able to operate in the market for products that are provided under those contracts. Additionally, if we are unable to consistently win new contract awards over an extended period, or if we fail to anticipate all of the costs or resources that we will be required to secure and, if applicable, perform under such contract awards, our growth strategy and our business, financial condition and operating results and cash flows could be materially and adversely affected.
There are a number of laws and regulations that pertain to government contracts and compliance with those laws and regulations require significant time and cost, which could have a material adverse effect on our business, financial condition, operating results and cash flows.
As a manufacturer and supplier of MCMs to the USG addressing PHTs, we must comply with numerous laws and regulations relating to the procurement, formation, administration and performance of government contracts. These laws and regulations govern how we transact business with our government clients and, in some instances, impose additional costs and related obligations on our business operations. Our status as a USG contractor means that we are subject to various statutes and regulations, including:
• the Federal Acquisition Regulation (FAR) and agency-specific regulations supplemental to FAR, which comprehensively regulate the award, formation, administration and performance of government contracts;
• the Defense Federal Acquisition Regulations (DFARs) and agency-specific regulations supplemental to DFARs, which comprehensively regulate the award, formation, administration and performance of DoD government contracts;
• the Department of State Acquisition Regulation (DOSAR) which regulates the relationship between a Department of State organization and a contractor or potential contractor;
• business ethics and public integrity obligations, which govern conflicts of interest and the hiring of former government employees, restrict the granting of gratuities and funding of lobbying activities and incorporate other requirements such as the Anti-Kickback Act, the Procurement Integrity Act, the False Claims Act and the Foreign Corrupt Practices Act;
• export and import control laws and regulations, including but not limited to ITAR (International Traffic in Arms Regulations); and
• laws, regulations and executive orders restricting the use and dissemination of information classified for national security purposes and the exportation of certain products and technical data.
We may be subject to government investigations of compliance with government acquisition regulations. USG agencies routinely audit and investigate government contractors for compliance with applicable laws and standards. Even though we take significant precautions to identify, prevent and deter fraud, misconduct and non-compliance, we face the risk that our personnel or outside partners may engage in misconduct, fraud or improper activities. If we are audited or investigated and such audit or investigation were to uncover improper or illegal activities, we could be subject to civil and criminal fines and penalties, administrative sanctions, including suspension or debarment from government contracting, and suffer significant reputational harm. The loss of our status as an eligible government contractor or significant fines or penalties associated with contract non-compliance or resulting from investigations could have a material adverse effect on our business.
The amount we are paid under our fixed price government procurement contracts is based on estimates we have made of the time, resources and expenses required for us to perform under those contracts. If our actual costs exceed our estimates, we may not be able to earn an adequate return or may incur a loss under these contracts, which could harm our operating results and materially reduce our net income.
Our current procurement contracts with HHS and DoD are generally fixed price contracts. We expect that additional future procurement contracts we successfully secure with the USG would likely also be fixed price contracts. Under a fixed price contract, we are required to deliver our products at a fixed price regardless of the actual costs we incur. Estimating costs that are related to performance in accordance with contract specifications is difficult, particularly where the period of performance is over several years. Our failure to anticipate technical problems, estimate costs accurately or control costs during performance of a fixed price contract could reduce the profitability of such a contract or cause a loss, which could harm our operating results and materially reduce our net income.
Unfavorable provisions in government contracts, some of which may be customary, may subject our business to material limitations, restrictions and uncertainties and may have a material adverse impact on our business, financial condition, operating results and cash flows.
Government contracts customarily contain provisions that give the USG substantial rights and
42
EMERGENT BIOSOLUTIONS INC.
remedies, many of which are not typically found in commercial contracts, including provisions that allow the USG to:
•terminate existing contracts, in whole or in part, for any reason;
•unilaterally reduce or modify contracts or subcontracts;
•decline, in whole or in part, to exercise an option to purchase product under a procurement contract or to fund additional development under a development contract;
•decline to renew a procurement contract;
•claim certain rights to facilities or to products, including intellectual property, developed under the contract;
•require repayment of contract funds spent on construction of facilities in the event of contract default;
•take actions that result in a longer development timeline than expected;
•direct the course of a development program in a manner not chosen by the government contractor;
•suspend or debar the contractor from doing business with the government or a specific government agency;
•pursue civil or criminal remedies under acts such as the False Claims Act and False Statements Act; and
•control or prohibit the export of products.
Generally, government contracts contain provisions permitting unilateral termination or modification, in whole or in part, at the USG’s convenience. Under general principles of government contracting law, if the USG terminates a contract for convenience, the government contractor may recover only its incurred or committed costs, settlement expenses and profit on work completed prior to the termination. If the USG terminates a contract for default, the government contractor is entitled to recover costs incurred and associated profits on accepted items only and may be liable for excess costs incurred by the government in procuring undelivered items from another source. All of our development and procurement contracts with the USG are terminable at the USG's convenience with these potential consequences.
In addition, our USG contracts grant the USG the right to use technologies developed by us under the government contract or the right to share data related to our technologies, for or on behalf of the USG. Under
our USG contracts, we may not be able to limit third parties, including our competitors, from accessing certain of these technology or data rights, including intellectual property, in providing products and services to the USG.
PRODUCT DEVELOPMENT AND COMMERCIALIZATION RISKS
An inability to maintain quality and manufacturing compliance at our manufacturing facilities could hinder our ability to continue producing bulk drug substance for Johnson & Johnson’s COVID-19 vaccine and the products and product candidates of other CDMO customers, which could adversely affect our business, financial condition, operating results and cash flows.
The FDA conducts periodic inspections of our manufacturing facilities for compliance with cGMP requirements relating to quality control. The potential failure to maintain compliance with such standards at our manufacturing facilities could hinder our ability to continue manufacturing for CDMO customers. For example, in April 2021, the FDA conducted an inspection of our Baltimore Bayview facility after an out-of-specification result was discovered involving the cross-contamination of a single drug substance lot intended for further drug product manufacturing and use in Johnson & Johnson’s COVID-19 vaccine. The inspection revealed, among other things, the need for a more thorough investigation to explain cross-contamination issues identified in a viral vaccine drug substance batch intended for use in Johnson and Johnson’s COVID-19 vaccine, and that improved building maintenance and sanitation practices were needed at the Bayview facility, which resulted in the issuance of a Form FDA 483, the temporary suspension of production of the bulk drug substance for Johnson & Johnson’s COVID-19 vaccine and the removal of production related to AstraZeneca’s COVID-19 from Bayview. We are currently in negotiations to settle the remainder of our contract with AstraZeneca to focus solely on producing Johnson & Johnson’s bulk drug substance at Bayview. Additional failures to maintain compliance with cGMP requirements relating to quality control at our manufacturing facilities could hinder our ability to continue manufacturing for our CDMO customers, including the bulk drug substance for Johnson & Johnson’s COVID-19 vaccine, which could adversely affect our business, financial condition, operating results and cash flows.
The COVID-19 product candidates we are working on may not be safe or effective and, even if they are, we may not be able to manufacture sufficient quantities to meet demand.
43
EMERGENT BIOSOLUTIONS INC.
We are developing a product candidate for the possible treatment of COVID-19 in the outpatient setting and we are also providing CDMO services for the development and/or manufacture of multiple vaccine and therapeutic product candidates. There can be no assurance that any of these product candidates will be safe or effective. There can also be no assurance that any of these product candidates will be authorized for emergency use or approved by the FDA or any other health regulatory authority or that our facilities will receive authorization from the FDA to release additional batches of COVID-19 drug substance. Even if these product candidates are safe and/or effective and receive authorization or approval by a health regulatory authority or we receive authorization to produce drug substance at our facilities, the manufacturing processes for our CDMO COVID-19 programs are under development and are complex. There can be no assurance that we will be able to produce any significant quantity of these product candidates in a timely basis or at all, or negotiate further commitments under our existing CDMO contracts to manufacture vaccines against COVID-19, which could adversely affect our business, financial condition, operating results and cash flows.
Our growth depends on our success in developing and commercializing our product candidates. If we are unable to commercialize these product candidates, or experience significant delays or unanticipated costs in doing so, our business would be materially and adversely affected.
We have invested significant efforts and financial resources in the development of our vaccines, therapeutics and medical device product candidates and the acquisition of additional product candidates. In addition to our product sales, our ability to generate revenue is dependent on a number of factors, including the success of our development programs, the USG's interest in providing development funding for or procuring certain of our product candidates, and the commercial viability of our acquired or developed product candidates. The commercial success of our product candidates will depend on many factors, including accomplishing the following in an economical manner:
•successful development, formulation and cGMP scale-up of manufacturing that meets FDA or other foreign regulatory requirements;
•successful program partnering;
•successful completion of clinical or non-clinical development;
•receipt of marketing approvals from the FDA and equivalent foreign regulatory authorities;
•establishment of commercial manufacturing processes and product supply arrangements;
•training of a commercial sales force for the product;
•successful registration and maintenance of relevant patent and/or other proprietary protection; and
•acceptance of the product by potential government and other customers.
Clinical trials of product candidates are expensive and time-consuming, and their outcome is uncertain. We must invest substantial amounts of time and financial resources in these trials, which may not yield viable products. Failure to obtain regulatory approval for product candidates, particularly in the United States, could materially and adversely affect our financial resources, which would adversely affect our business, financial condition, operating results and cash flows.
Before obtaining regulatory approval for the marketing of our product candidates, we and our collaborative partners, where applicable, must conduct pre-clinical studies and clinical trials to establish proof of concept and demonstrate the safety and efficacy of our product candidates. Pre-clinical and clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. Success in pre-clinical testing and early clinical trials does not ensure that later clinical trials will be successful, and interim results of such trials do not necessarily predict final results. An unexpected result in one or more of our clinical trials can occur at any stage of testing.
Pre-clinical and clinical testing for certain of our product candidates addressing CBRNE threats may face additional difficulties and uncertainties because they cannot ethically or feasibly be tested in human subjects. We therefore expect to rely on the Animal Rule to obtain regulatory approval for some of our CBRNE product candidates. The Animal Rule permits, in certain limited circumstances, the use of animal efficacy studies, together with human clinical safety and immunogenicity trials, to support an application for marketing approval. For a product approved under the Animal Rule, certain additional post-marketing requirements apply. For example, to the extent feasible and ethical, applicants must conduct post-marketing studies, such as field studies, to verify and describe the drug's clinical benefit and to assess its safety when used as indicated. It is possible that results from these animal efficacy studies may not be predictive of the actual efficacy of our product candidates in humans.
Prior to FDA approval of the countermeasure product candidates, the Secretary of HHS can contract to purchase MCMs for the SNS under Project BioShield under certain circumstances. Under PAHPRA, the USG may also, at its discretion, purchase critical biodefense
44
EMERGENT BIOSOLUTIONS INC.
products for the SNS prior to FDA approval after the filing of a pre-EUA application with the FDA. If our product candidates are not procured or funded under regulatory authority, they generally will have to be fully approved by the FDA through traditional regulatory mechanisms for distribution in the United States.
We may experience unforeseen events or issues during, or as a result of, pre-clinical testing, clinical trials or animal efficacy studies. These issues and events, which could delay or prevent our ability to receive regulatory approval for a product candidate, include, among others:
•our inability to manufacture sufficient quantities for use in trials;
•the unavailability or variability in the number and types of subjects for each study;
•safety issues or inconclusive or incomplete testing, trial or study results;
•drug immunogenicity;
•lack of efficacy of product candidates during the trials;
•government or regulatory restrictions or delays; and
•greater than anticipated costs of trials.
We may fail to select or capitalize on the most scientifically, clinically or commercially promising or profitable product candidates.
We continue to evaluate our product development strategy and, as a result, may modify our strategy in the future. In this regard, we may, from time to time, focus our product development efforts on different product candidates or may delay or halt the development of various product candidates. We may change or refocus our existing product development, commercialization and manufacturing activities based on government funding decisions. This could require changes in our facilities and our personnel. Any product development changes that we implement may not be successful. In particular, we may fail to select or capitalize on the most scientifically, clinically or commercially promising or profitable product candidates or choose candidates for which government development funds are not available. Our decisions to allocate our research and development, management and financial resources toward particular product candidates or therapeutic areas may not lead to the development of viable commercial products and may divert resources from better business opportunities. Similarly, our decisions to delay or terminate product development programs may also prove to be incorrect and could cause us to miss valuable opportunities.
GLOBAL PANDEMIC RISK
The COVID-19 coronavirus pandemic could have a material adverse impact on our business, results of operations and financial performance.
Our business, operations and financial condition and results have been and may continue to be impacted by the COVID-19 pandemic to varying degrees. The pandemic has presented a number of risks and challenges for our business, including, among others, government-mandated work-from-home or shelter-in-place orders; manufacturing disruptions and delays, including at our Baltimore Bayview facility, supply chain interruptions, including challenges related to reliance on third-party suppliers; disruptions to pipeline development and clinical trials and decreased product demand for our travel health vaccines due to the significant reduction in international travel. Additional travel restrictions and other governmental measures may result in further disruptions or continued delays in delivery of supplies by our third-party contractors and suppliers.
A significant number of our administrative employees continue to work from home due to policies implemented as a result of COVID-19. Working remotely could increase our cybersecurity risk, create data accessibility concerns, and make us more susceptible to communication disruptions, any of which could adversely impact our business operations. In addition, our on-site staff conducting research and development may not be able to access our laboratories if conditions worsen, due to state and local restrictions, and these core activities may be significantly limited or curtailed, possibly for extended periods of time.
We also face uncertainties related to our efforts and those of our collaborative partners to develop a potential treatment or vaccine for COVID-19, including uncertainties related to pre-clinical or clinical trials, the risk that such development programs may not be successful, commercially viable, or that EUA or regulatory approval will not be received from regulatory authorities.
In addition, the trading price of our common stock, and that of other biopharmaceutical companies, has been highly volatile due to the COVID-19 pandemic, especially as a result of investor concerns and uncertainty related to the impact of the pandemic on the economies of countries worldwide. These broad market and industry fluctuations, as well as general economic, political and market conditions, may negatively impact the market price of shares of our common stock.
The COVID-19 pandemic continues to rapidly evolve. The extent to which the pandemic and variants of COVID-19 further negatively impact our business,
45
EMERGENT BIOSOLUTIONS INC.
supply chain, disrupt key clinical trials, divert government funding away from our primary procured products and product candidates due to changes in government priorities and potential delays in the delivery of products to our customers will depend on future developments, which are highly uncertain. The ultimate geographic spread of COVID-19 and new variants of the disease, the duration of the pandemic, further travel restrictions and social distancing in the United States and other countries, business closures or business disruptions and the effectiveness of actions taken in the United States and other countries to contain and treat the disease cannot be predicted with certainty.
The continually evolving nature of the COVID-19 pandemic and the resulting public health response, including the changing demand for various COVID-19 vaccines and treatments from both patients and governments around the world, may affect sales of the COVID-19 product candidates manufactured by our CDMO business.
Through our CDMO business, we manufacture or provide services for a variety of product candidates intended for the prevention or treatment of COVID-19 and its symptoms and effects, including development services, the manufacture of bulk drug substance and fill and finish services.
All of the COVID-19-related product candidates we develop and manufacture have yet to receive full regulatory approval from any regulatory authority, although some are being marketed and sold pursuant to an EUA from the FDA or the equivalent authorization from non-U.S. regulatory authorities. Should the facilities producing these product candidates be denied an EUA or one or more of these COVID-19-related product candidates be denied an EUA (or equivalent) or be denied full regulatory approval by the FDA or other major non-U.S. regulatory authority, the demand for such product candidates could decrease significantly and therefore decrease customer orders for additional CDMO services for such product candidates. Additionally, the need for continued manufacture and supply of vaccines (including potential “booster” doses) and therapies to address the COVID-19 pandemic, including new and developing variants of COVID-19, is highly uncertain and subject to various political, economic and regulatory factors that are outside of our control. Should the U.S. or other major regions worldwide determine that additional manufacturing of COVID-19 vaccines, boosters, or therapies is no longer necessary, it could adversely affect our revenue and financial condition and our ability to grow our CDMO business in the near term. In addition, highly-public political and social debate relating to the need for, efficacy of, or side effects
related to one or more specific COVID-19 vaccines could contribute to changes in public perception of COVID-19 vaccines manufactured by us, which could decrease demand for a COVID-19 related product candidate we develop, manufacture (in whole or in part).
REGULATORY AND COMPLIANCE RISKS
Our long-term success depends, in part, upon our ability to develop, receive regulatory approval for and commercialize product candidates we develop or acquire and, if we are not successful, our business, financial condition, operating results and cash flows may suffer.
Our product candidates and the activities associated with them are subject to extensive FDA regulation and oversight, as well as oversight by other regulatory agencies in the United States and by comparable authorities in other countries. This includes, but is not limited to, laws and regulations governing product development, including testing, manufacturing, record keeping, storage and approval, as well as advertising and promotion. In limited circumstances, governments may procure products that have not obtained regulatory approval. In all other circumstances, failure to obtain regulatory approval for a product candidate will prevent us from selling and commercializing the product candidate.
In the United States, to obtain approval from the FDA to market any of our future drug, biologic, or vaccine products, we will be required to submit an NDA or BLA to the FDA. Ordinarily, the FDA requires a company to support an NDA or BLA with substantial evidence of the product candidate’s effectiveness, safety, purity and potency in treating the targeted indication based on data derived from adequate and well-controlled clinical trials, including Phase 3 trials conducted in patients with the disease or condition being targeted.
However, many of our MCM product candidates, for example, may take advantage of a different regulatory approval pathway under the FDA’s “Animal Rule.” Under the Animal Rule, efficacy must be demonstrated, in part, by utilizing animal models rather than testing in humans. We cannot guarantee that the FDA will permit us to proceed with licensure of any of our PHT MCM candidates under the Animal Rule. Even if we are able to proceed under the Animal Rule, product development can take a considerable amount of time, and the FDA may decide that our data are insufficient to support approval and require additional pre-clinical, clinical or other studies, refuse to approve our products, or place restrictions on our ability to commercialize those products. Furthermore, products approved under the Animal Rule are subject to certain additional post-marketing requirements. We cannot
46
EMERGENT BIOSOLUTIONS INC.
guarantee that we will be able to meet this regulatory requirement even if one or more of our product candidates are approved under the Animal Rule.
The process of obtaining these regulatory approvals is expensive, often takes many years if approval is obtained at all, and can vary substantially based upon the type, complexity and novelty of the product candidate involved. Changes in the regulatory approval process may cause delays in the approval or rejection of an application. There is a high rate of failure inherent in this process, and potential products that appear promising at early stages of development may fail for a number of reasons, and positive results from pre-clinical studies may not be predictive of similar results in human clinical trials. Similarly, promising results from earlier clinical trials of a product candidate may not be replicated in later clinical trials.
There are many other difficulties and uncertainties inherent in pharmaceutical research and development that could significantly delay or otherwise materially delay our ability to develop future product candidates, mostly related to clinical trials.
Failure to successfully develop future product candidates may materially adversely affect our business, financial condition, operating results and cash flows.
Once an NDA or BLA is submitted, the FDA has substantial discretion and may refuse to accept any application or may decide that our data are insufficient to support approval and require additional pre-clinical, clinical or other studies.
Unapproved and investigational stage products are also subject to the FDA's laws and regulations governing advertising and promotion, which prohibit the promotion of both unapproved products and unapproved uses of approved products. There is some risk that the FDA could conclude that our communications relating to unapproved products or unapproved uses of approved products constitute the promotion of an unapproved product or product use in violation of FDA laws and regulations. There is also a risk that a regulatory authority in another country could take a similar position under that country's laws and regulations and conclude that we have violated the laws and regulations related to product development, approval, or promotion in that country. Therefore, there is a risk that we could be subject to enforcement actions if found to be in violation of such laws or regulations.
Even if we or our collaborators obtain marketing approvals for our product candidates, the conditions of approvals and ongoing regulation of our products may limit how we manufacture and market our products, which could materially impair our ability to generate revenue.
Once approval has been granted, an approved product and its manufacturer and marketer remain subject to ongoing review and extensive regulation.
We and our collaborators must therefore comply with requirements concerning advertising and promotion for any of our product candidates for which we obtain marketing approval. Promotional communications with respect to FDA-regulated products are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved labeling. Thus, we will not be able to sell any products we develop for indications or uses for which they are not approved.
If we and our collaborators are not able to comply with post-approval regulatory requirements, we could have the marketing approvals for our products withdrawn by regulatory authorities and our ability to market any products could be limited, which could adversely affect our ability to achieve or sustain profitability. Further, the cost of compliance with post-approval regulations may have a negative effect on our operating results and financial condition.
Any product candidate for which we or our collaborators obtain marketing approval could be subject to restrictions or withdrawal from the market and we may be subject to substantial penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with our product candidates, when and if any of them are approved.
Any product candidate for which we or our collaborators obtain marketing approval, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for such product, will be subject to continual requirements of and review by the FDA and other regulatory authorities. These requirements include submissions of safety and other post-marketing information and reports, registration and listing requirements, cGMP requirements relating to quality control and manufacturing, quality assurance and corresponding maintenance of records and documents, and requirements regarding the distribution of samples to physicians and recordkeeping. Even if marketing approval of a product candidate is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the medicine.
Certain of our products are subject to post marketing requirements (PMRs), which we are required to conduct, and post marketing commitments (PMCs), which we have agreed to conduct. The FDA has the authority to take action against sponsors who fail to meet the obligations of a PMR, including civil monetary penalties and/or misbranding charges.
47
EMERGENT BIOSOLUTIONS INC.
The FDA and other agencies, including the U.S. Department of Justice (DOJ) and the HHS Office of Inspector General (OIG), closely regulate and monitor the pre-approval and post-approval marketing and promotion of products to ensure that they are marketed and distributed only for the approved indications and in accordance with the provisions of the approved labeling. The FDA, DOJ, and OIG impose stringent restrictions on manufacturers’ communications regarding unapproved products and unapproved uses of approved products and if we market unapproved products or market our approved products for unapproved indications, we may be subject to enforcement action. Violations of the FDCA and other statutes, including the False Claims Act, relating to the promotion and advertising of prescription products may lead to investigations and enforcement actions alleging violations of federal and state health care fraud and abuse laws, as well as state consumer protection laws.
In addition, later discovery of previously unknown adverse events or other problems with our products, manufacturing partners or manufacturing processes, or failure to comply with regulatory requirements, may result in various penalties and sanctions. For all FDA-regulated products, if the FDA finds that a manufacturer has failed to comply with applicable laws and regulations, or that a product is ineffective or poses an unreasonable health risk, it can institute or seek a wide variety of enforcement actions and remedies, including but not limited to:
• restrictions on such products, manufacturers or manufacturing processes;
• restrictions on the labeling or marketing of a product;
• restrictions on distribution or use of a product;
• requirements to conduct post-marketing studies or clinical trials;
• warning letters or untitled letters;
• refusal to approve pending applications or supplements to approved applications that are submitted;
• fines, restitution or disgorgement of profits or revenues;
• suspension or withdrawal of marketing approvals;
• refusal to permit the import or export of our products;
• product seizure; and
• injunctions or the imposition of civil or criminal penalties.
Non-compliance with EU requirements regarding safety monitoring or pharmacovigilance, and with requirements related to the development of products for the pediatric population, can also result in significant financial penalties. Similarly, failure to
comply with the EU and other legal and regulatory requirements regarding the protection of personal information can also lead to significant penalties and sanctions. Non-compliance with similar requirements in other foreign jurisdictions can also result in enforcement actions and significant penalties.
Current and future legislation may increase the difficulty and cost for us and any collaborators to obtain marketing approval of and commercialize our product candidates and affect the prices we, or they, may obtain.
In the United States and foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the health care system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product candidates for which we obtain marketing approval. We expect that current laws, as well as other health care reform measures that may be adopted in the future, may result in more rigorous coverage criteria and additional downward pressure on the price that we, or any collaborators, may receive for any approved products.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, the ACA), passed in 2010 substantially changed the way health care is financed by both governmental and private insurers, and significantly impacted the U.S. biopharmaceutical industry. However, some provisions of the ACA have yet to be fully implemented and certain provisions have been subject to legal and political challenges, as well as efforts by the last Presidential administration to repeal or replace certain aspects of the ACA. More recently on January 28, 2021, however, the President issued an executive order to strengthen implementation of the ACA. Concurrently, Congress considered legislation that would repeal or repeal and replace all or part of the ACA. While Congress has not passed comprehensive repeal legislation, it has enacted laws that modify certain provisions of the ACA, such as removing penalties as of January 1, 2019 for not complying with the ACA’s individual mandate to carry health insurance, delaying the implementation of certain ACA-mandated fees, and increasing the point-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA. Prior to the Supreme Court’s decision, the current Presidential administration issued an executive order initiating a special enrollment period from February 15, 2021 through August 15, 2021 for purposes of obtaining health insurance coverage through the ACA
48
EMERGENT BIOSOLUTIONS INC.
marketplace. The executive order also instructed certain governmental agencies to review and reconsider their existing policies and rules that limit access to healthcare. It is unclear how healthcare reform measures enacted by Congress or implemented by the current Presidential administration or other challenges to the ACA, if any, will impact the ACA or our business.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA was enacted that may negatively impact us. On August 2, 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions of Medicare payments to providers of up to 2% per fiscal year. These reductions went into effect on April 1, 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2030 under the CARES Act.
Additionally, there has been recent heightened federal governmental scrutiny over the manner in which manufacturers set prices for their marketed products. For example, there have been several recent Congressional inquiries and has been proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. For example, the last Presidential administration released a “Blueprint”, or plan, to lower drug prices and reduce out of pocket costs of drugs that contains additional proposals to increase drug manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products, and reduce the out-of-pocket costs of drug products paid by consumers.
At the state level, individual states are increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition, regional health care authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will
be included in their prescription drug and other health care programs. These measures could reduce the ultimate demand for our products, once approved, or put pressure on our product pricing. We expect that additional state and federal health care reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for health care products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
If we fail to comply with foreign, federal, state and local health care laws, including fraud and abuse and health information privacy and security laws, and antitrust laws, we could face substantial penalties and our business, results of operations, financial condition and prospects could be adversely affected.
In the United States, certain of our products are reimbursed under federal and state health care programs such as Medicaid, Medicare, TriCare, and/or state pharmaceutical assistance programs. Many foreign countries have similar laws. Federal and state laws designed to prevent fraud and abuse under these programs prohibit pharmaceutical companies from offering valuable items or services to customers or potential customers to induce them to buy, prescribe, or recommend our product (the so-called “anti-kickback” laws). Exceptions are provided for discounts and certain other arrangements if specified requirements are met. Other federal and state laws, and similar foreign laws, not only prohibit us from submitting any false information to government reimbursement programs but also prohibit us, our employees, or any third party acting on our behalf from doing anything to cause, assist, or encourage our customers to submit false claims for payment to these programs. We are also subject to various federal, state and foreign antitrust and competition laws that prohibit certain activities that may have an impact against potential competitors. Violations of the various fraud and abuse and antitrust laws may result in severe penalties against the responsible employees and us, including jail sentences, large fines, and the exclusion of our products from reimbursement under federal and state programs. Some of the laws that may affect our ability to operate include:
•the federal Anti-Kickback Statute makes it illegal for any person or entity, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, receive, offer or pay remuneration, directly or indirectly, overtly or covertly, to induce, or in return for, either the referral of an individual, or the purchase, lease, prescribing or recommendation of an item, good, facility or service reimbursable by a federally funded health care program, such as the Medicare or Medicaid program. The term “remuneration”
49
EMERGENT BIOSOLUTIONS INC.
has been interpreted broadly and may constrain our marketing practices, educational programs, pricing policies and relationships with health care providers or other entities, among other activities;
•the federal False Claims Act imposes criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for, among other things, knowingly presenting, or causing to be presented, false or fraudulent claims for payment by a federal health care program or making a false statement or record material to payment of a false claim or avoiding, decreasing or concealing an obligation to pay money to the federal government, with potential liability, including mandatory treble damages and significant per-claim penalties.
•the U.S. federal Health Insurance Portability and Accountability Act of 1996 (HIPAA), which imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any health care benefit program or obtain, by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any health care benefit program, regardless of the payor (e.g., public or private) and knowingly and willfully falsifying, concealing or covering up by any trick or device a material fact or making any materially false statement, in connection with the delivery of, or payment for, health care benefits, items or services. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation;
•HIPAA, as amended by HITECH, and their respective implementing regulations mandates, among other things, the adoption of uniform standards for the electronic exchange of information in common health care transactions, as well as standards relating to the privacy, security and transmission of individually identifiable health information, which require the adoption of administrative, physical and technical safeguards to protect such information. Among other things, HITECH makes HIPAA's security standards directly applicable to “business associates,” or independent contractors or agents of covered entities that create, receive or obtain protected health information in connection with providing a service for or on behalf of a covered entity;
•the Physician Payments Sunshine Act and its implementing regulations require certain manufacturers of drugs, biologics, medical devices and medical supplies for which payment is available under Medicare, Medicaid or the Centers for Medicare & Medicaid Services (CMS) to report certain payments and transfers of value made to U.S. physicians and teaching hospitals, and ownership or investment interests held by physicians and their immediate family members. Beginning in 2022, applicable manufacturers will also be required to report information regarding payments and transfers of value provided to U.S. physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, and certified nurse-midwives; and
•state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third-party payor, including commercial insurers; state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; state, local and foreign laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, obtain pharmaceutical agent licensure, and/or otherwise restrict payments that may be made to health care providers and entities; and state, local and foreign laws that require drug manufacturers to report information related to payments and other transfers of value to health care providers or entities, or marketing expenditures.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available under the federal Anti-Kickback Statute, it is possible that some of our business activities could be subject to challenges under one or more of such laws. Moreover, recent health care reform legislation has strengthened these laws. For example, the ACA, among other things, amends the intent requirement of the federal Anti-Kickback Statute and criminal health care fraud statutes, so that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it. In addition, the ACA provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act.
50
EMERGENT BIOSOLUTIONS INC.
If our operations are found to be in violation of any of the laws described above or otherwise, we may be subject to penalties, including civil and criminal penalties, damages, fines, individual imprisonment, integrity obligations, exclusion from funded health care programs and the curtailment or restructuring of our operations. Any such penalties could adversely affect our financial results. We continue to improve our corporate compliance program designed to ensure that our development, marketing, and sales of existing and future products and product candidates are in compliance with all applicable laws and regulations, but we cannot guarantee that this program will protect us from governmental investigations or other actions or lawsuits stemming from a failure to comply with such laws or regulations. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Efforts to ensure that our business arrangements with third parties will comply with health care laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving fraud and abuse or other health care laws and regulations. If our operations are found to be in violation of any of these laws, we may be subject to significant civil, criminal and administrative penalties, damages, fines, individual imprisonment, integrity obligations, exclusion from government funded health care programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations. If a third party fails to comply with applicable laws and regulations while acting on our behalf, we may also be subject to criminal, civil, and administrative penalties, including those listed above.
We are committed to conducting the development, sale and marketing of our applicable products and product candidates and all of our activities in compliance with all applicable laws and regulations, but certain applicable laws and regulations may impose liability even in the absence of specific intent to defraud. Furthermore, should an employee or third party acting on our behalf violate these laws without our knowledge, a governmental authority may impose civil and/or criminal sanctions on us.
The United States government, state governments and private payors regularly investigate the pricing and competitive practices of pharmaceutical companies and biotechnology companies, and many file actions alleging that inaccurate reporting of prices has improperly inflated reimbursement rates. We may also be subject to investigations related to our pricing practices. Regardless of merit or eventual outcome,
these types of investigations and related litigation can result in:
•Diversion of management time and attention;
•Significant legal fees and payment of damages or penalties;
•Limitations on our ability to continue certain operations;
•Decreased product demand; and
•Injury to our reputation.
Moreover, an adverse outcome, or the imposition of penalties or sanctions for failing to comply with the fraud and abuse and antitrust laws, could adversely affect us and may have a material adverse effect on our business, results of operations, financial condition and cash flows.
If we fail to comply with our obligations under U.S. governmental pricing programs, we could be required to reimburse government programs for underpayments and could pay penalties, sanctions and fines.
The issuance of regulations and coverage expansion by various governmental agencies relating to the Medicaid rebate program will continue to increase our costs and the complexity of compliance and will be time-consuming. Changes to the definition of “average manufacturer price” (AMP), and the Medicaid rebate amount under the ACA and CMS and the issuance of final regulations implementing those changes has affected and could further affect our 340B “ceiling price” calculations. Because we participate in the Medicaid rebate program, we are required to report “average sales price” (ASP), information to CMS for certain categories of drugs that are paid for under Part B of the Medicare program. Future statutory or regulatory changes or CMS binding guidance could affect the ASP calculations for our products and the resulting Medicare payment rate and could negatively impact our results of operations.
Pricing and rebate calculations vary among products and programs, involve complex calculations and are often subject to interpretation by us, governmental or regulatory agencies and the courts. The Medicaid rebate amount is computed each quarter based on our submission to CMS of our current AMP and “best price” for the quarter. If we become aware that our reporting for a prior quarter was incorrect, or has changed as a result of recalculation of the pricing data, we are obligated to resubmit the corrected data for a period not to exceed twelve quarters from the quarter in which the data originally were due. Any such revisions could have the impact of increasing or decreasing our rebate liability for prior quarters, depending on the direction of the revision. Such restatements and recalculations would increase our
51
EMERGENT BIOSOLUTIONS INC.
costs for complying with the laws and regulations governing the Medicaid rebate program. Price recalculations also may affect the “ceiling price” at which we are required to offer our products to certain covered entities, such as safety-net providers, under the 340B/Public Health Service (PHS) drug pricing program.
In addition, if we are found to have made a misrepresentation in the reporting of ASP, we are subject to civil monetary penalties for each such price misrepresentation and for each day in which such price misrepresentation was applied. If we are found to have knowingly submitted false AMP or “best price” information to the government, we may be liable for civil monetary penalties per item of false information. Any refusal of a request for information or knowing provision of false information in connection with an AMP survey verification would also subject us to civil monetary penalties. In addition, our failure to submit monthly/quarterly AMP or “best price” information on a timely basis could result in a civil monetary penalty per day for each day the information is late beyond the due date. Such failure also could also be grounds for CMS to terminate our Medicaid drug rebate agreement, under which we participate in the Medicaid program. In the event that CMS terminates our rebate agreement, no federal payments would be available under Medicaid or Medicare Part B for our covered outpatient drugs. Governmental agencies may also make changes in program interpretations, requirements or conditions of participation, some of which may have implications for amounts previously estimated or paid. We cannot assure that our submissions will not be found by CMS to be incomplete or incorrect.
In order for our products to be reimbursed by the primary federal governmental programs, we must report certain pricing data to the USG. Compliance with reporting and other requirements of these federal programs is a pre-condition to: (i) the availability of federal funds to pay for our products under Medicaid and Medicare Part B; and (ii) procurement of our products by the Department of Veterans Affairs (DVA), and by covered entities under the 340B/PHS program. The pricing data reported are used as the basis for establishing Federal Supply Schedule (FSS), and 340B/PHS program contract pricing and payment and rebate rates under the Medicare Part B and Medicaid programs, respectively. Pharmaceutical companies have been prosecuted under federal and state false claims laws for submitting inaccurate and/or incomplete pricing information to the government that resulted in increased payments made by these programs. Although we maintain and follow strict procedures to ensure the maximum possible integrity for our federal pricing calculations, the process for making the required calculations is complex, involves some subjective judgments and the risk of errors
always exists, which creates the potential for exposure under the false claims laws. If we become subject to investigations or other inquiries concerning our compliance with price reporting laws and regulations, and our methodologies for calculating federal prices are found to include flaws or to have been incorrectly applied, we could be required to pay or be subject to additional reimbursements, penalties, sanctions or fines, which could have a material adverse effect on our business, financial condition and results of operations.
To be eligible to have our products paid for with federal funds under the Medicaid and Medicare Part B programs and purchased by certain federal agencies and grantees, we also must participate in the DVA FSS pricing program. To participate, we are required to enter into an FSS contract with the DVA, under which we must make our innovator “covered drugs” available to the “Big Four” federal agencies-the DVA, the DoD, the Public Health Service (including the Indian Health Service), and the Coast Guard-at pricing that is capped under a statutory federal ceiling price (FCP) formula set forth in Section 603 of the Veterans Health Care Act of 1992 (VHCA). The FCP is based on a weighted average wholesale price known as the Non-Federal Average Manufacturer Price (Non-FAMP), which manufacturers are required to report on a quarterly and annual basis to the DVA. Under the VHCA, knowingly providing false information in connection with a Non-FAMP filing can subject us to significant penalties for each item of false information. If we overcharge the government in connection with our FSS contract or Section 703 Agreement, whether due to a misstated FCP or otherwise, we are required to disclose the error and refund the difference to the government. The failure to make necessary disclosures and/or to identify contract overcharges can result in allegations against us under the False Claims Act and other laws and regulations. Unexpected refunds to the government, and responding to a government investigation or enforcement action, can be expensive and time-consuming, and could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
From time to time, we sell unapproved MCMs to government entities under certain circumstances. While this is permissible in some cases, the extent to which we may be able to lawfully offer to sell and sell unapproved products in many jurisdictions may be unclear or ambiguous. Such sales could subject us to regulatory enforcement action, product liability and reputational risk.
Under certain circumstances, MCMs may be procured by government entities prior to approval by the FDA or other regulatory authorities, a practice which we follow in connection with AV7909 and Trobigard. In the United States, Project BioShield permits the
52
EMERGENT BIOSOLUTIONS INC.
Secretary of HHS to contract to purchase MCMs for the SNS prior to FDA approval of the countermeasure in specified circumstances. Project BioShield and the Pandemic and All-Hazards Preparedness Reauthorization Act of 2013 also allow the FDA Commissioner to authorize the emergency use of medical products that have not yet been approved by the FDA under an EUA. An EUA terminates when the emergency determination underlying the EUA terminates. An EUA is not a long-term alternative to obtaining FDA approval, licensure, or clearance for a product. Absent an applicable exception, our MCM product candidates generally will have to be approved by the FDA or other regulatory authorities in the relevant country through traditional pathways before we can sell those products to governments. Additionally, the laws in certain jurisdictions regarding the ability of government entities to purchase unapproved product candidates are ambiguous, and the permissibility of exporting unapproved products from the United States and importing them to foreign countries may be unclear. Nevertheless, government bodies, such as U.S. federal entities other than HHS, state and local governments within the United States, and foreign governments have sought and may further seek to procure our MCM product candidates that are not yet approved. If so, we would expect to assess the permissibility and liability implications of supplying our product candidates to such entities on a case-by-case basis, which presents certain challenges, both in the case of U.S. and foreign governments, and particularly under emergency conditions. In addition, agencies or branches of one country’s government may take different positions regarding the permissibility of such sales than another country’s government or even other agencies or branches of the same government. If local enforcement authorities disagree with our conclusion that such activities are permissible, they may take enforcement action against us.
In addition, the sale of unapproved products also could give rise to product liability claims for which we may not be able to obtain indemnification or insurance coverage. For example, liability protections applicable to claims arising under U.S. law and resulting from the use of certain unlicensed or unauthorized products, such as a declaration issued under the PREP Act, may lead plaintiffs to assert that their claims are not barred under the PREP Act.
Regardless of the permissibility and liability risks, in the event a user of one or more of our products suffers an adverse event, we may be subject to additional reputational risk if the product has not been approved by the FDA or the corresponding regulatory authority of another country, particularly because we will not have approved labeling regarding the safety or efficacy of those products. In addition, legislatures and other governmental bodies that have oversight
responsibility for procuring agencies may raise concerns after the fact, even if procurement was permissible at the time, which could result in negative publicity, reputational risk and harm to our business prospects.
There is also a risk that our communications with governments about our unapproved products, such as in the procurement context, could be considered promotion of an unapproved product or unapproved use of an approved product. Therefore, there is a risk that we could be subject to enforcement actions if found to be in violation of such laws or regulations.
Even after regulatory approval is received, if we fail to comply with regulatory requirements, or if we experience unanticipated problems with our approved products, they could be subject to restrictions, penalties or withdrawal from the market.
In addition to the requirements and uncertainties related to the pre-approval activities discussed previously, any vaccine, therapeutic product or medical device for which we obtain marketing approval, along with the manufacturing processes, post-approval clinical data, labeling, advertising and promotional activities for such product, will be subject to the continual requirements of and review by the FDA and other regulatory bodies. Our approved products are subject to these requirements and ongoing review. These requirements include submissions of safety and other post-marketing information and reports, plasma donor testing, registration requirements, cGMP, requirements relating to potency and stability, quality control, quality assurance, restrictions on advertising and promotion, import and export restrictions and recordkeeping requirements. In addition, various state laws require that companies that manufacture and/or distribute drug products within the state obtain and maintain a manufacturer or distributor license, as appropriate. Because of the breadth of these laws, it is possible that some of our business activities could be subject to challenge under one or more of such laws.
Government regulators enforce cGMP and other requirements through periodic unannounced inspections of manufacturing facilities. The FDA is authorized to inspect domestic and foreign manufacturing facilities without prior notice at reasonable times and in a reasonable manner. Health Canada may conduct similar inspections of our domestic and foreign facilities where Canadian marketed products are produced, or related formulation and filling operations are conducted. The FDA, Health Canada, and other foreign regulatory agencies conduct periodic inspections of our facilities. Following several of these inspections, regulatory authorities have issued inspectional observations, some of which were significant, but all of which are being, or have been, addressed through corrective actions. If, in connection
53
EMERGENT BIOSOLUTIONS INC.
with any future inspection, regulatory authorities find that we are not in substantial compliance with all applicable requirements, or if they are not satisfied with the corrective actions we take, our regulators may undertake enforcement action against us, which may include:
•warning letters and other communications;
•product seizure or withdrawal of the product from the market;
•restrictions on the marketing or manufacturing of a product;
•suspension or withdrawal of regulatory approvals or refusal to approve pending applications or supplements to approved applications;
•fines or disgorgement of profits or revenue; and
•injunctions or the imposition of civil or criminal penalties.
Similar action may be taken against us should we fail to comply with regulatory requirements, or later discover previously unknown problems with our products or manufacturing processes. For instance, our products are tested regularly to determine if they satisfy potency and stability requirements for their required shelf lives. Failure to meet potency, stability or other specification requirements could result in delays in distributions, recalls or other consequences. Even if regulatory approval of a product is granted, the approval may be subject to limitations on the indicated uses for which the product may be marketed or to the conditions of approval. Regulatory approval may also contain requirements for costly post-marketing testing and surveillance to monitor the safety or efficacy of the product. If we experience any of these post-approval events, our business, financial condition, operating results and cash flows could be materially and adversely affected.
Additionally, companies may not promote unapproved products or unapproved uses of approved products (i.e. “off-label” uses or uses that are not described in the product’s approved labeling and that differ from the uses approved by the applicable regulatory agencies). A company that is found to have improperly promoted an unapproved product or unapproved use of an approved product may be subject to significant liability, including civil and administrative remedies (such as entering into corporate integrity agreements with the USG), as well as criminal sanctions. If our employees or agents engage in marketing of an unapproved product or the unapproved use of an approved product, we could be subject to
civil or criminal investigations and monetary and injunctive penalties, which could adversely impact our ability to conduct business in certain markets, negatively affect our business, financial condition, operating results and cash flows, and damage our reputation.
Failure to obtain or maintain regulatory approval in international jurisdictions could prevent us from marketing our products abroad and could limit the growth of our business.
We currently sell certain of our products outside the United States and intend to expand the countries in which we sell our products and have received market authorization under the mutual recognition procedure to sell BioThrax in France, Italy, the Netherlands, Poland, and the United Kingdom. To market our products in foreign jurisdictions under normal circumstances, we generally need to obtain separate regulatory approvals and comply with numerous and varying requirements or use alternative “emergency use” or other exemptions from general approval and import requirements. Approval by the FDA in the United States or the mutual recognition procedure in the European member states does not ensure approval by all foreign regulatory authorities. The approval procedures in foreign jurisdictions can vary widely and can involve additional clinical trials and data review beyond that required by the FDA or under the mutual recognition procedure. There is also a risk that a regulatory authority in another country could conclude that we have violated the rules and regulations related to product development, approval or promotion in that country. Therefore, there is a risk that we could be subject to a foreign enforcement action if found to be in violation of such laws and regulations. We and our collaborators may not be able to obtain foreign regulatory approvals on a timely basis, if at all, and we may be unable to successfully commercialize our products in desired jurisdictions internationally if no alternate procurement pathway is identified for authorized government customers in a particular jurisdiction. We have limited experience in preparing, filing and prosecuting the applications necessary to gain foreign regulatory approvals and expect to rely on third-party contract research organizations and consultants to assist us in this process. Our reliance on third parties can introduce additional uncertainty into the process.
On January 31, 2020, the United Kingdom formally withdrew from the European Union and entered into a transition period through December 31, 2020 under a withdrawal agreement. On December 24, 2020, the United Kingdom and European Union entered into a Trade and Cooperation Agreement to govern the United Kingdom’s departure from the European Union, known as Brexit. Since a significant proportion of the regulatory framework in the United Kingdom is derived
54
EMERGENT BIOSOLUTIONS INC.
from European Union directives and regulations, the effects of the U.K.'s departure from the E.U. could materially impact the regulatory regime with respect to the approval of our products or product candidates in the United Kingdom or the European Union. Any delay in obtaining, or an inability to obtain, any marketing approvals, as a result of Brexit or otherwise, would prevent us from commercializing product candidates in the United Kingdom and/or the European Union and could restrict our ability to generate revenue and achieve and sustain profitability. Therefore, there is a risk that we could be subject to an enforcement action if found to be in violation of such laws or regulations.
Laws and regulations governing international operations may preclude us from developing, manufacturing and selling certain products outside of the United States and require us to develop and implement costly compliance programs.
As we continue to expand our commercialization activities outside of the United States, we are subject to an increased risk of, and must dedicate additional resources towards avoiding inadvertently conducting activities in a manner that violates the Foreign Corrupt Practices Act (FCPA), the U.K. Bribery Act, Canada's Corruption of Foreign Public Officials Act, and other similar foreign laws, which prohibit corporations and individuals from paying, offering to pay, or authorizing the payment of anything of value to any foreign government official, government staff member, political party, or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity. The FCPA also obligates companies whose securities are listed in the United States to comply with certain accounting provisions requiring the Company to maintain books and records that accurately and fairly reflect all transactions of the corporation, including international subsidiaries, and to devise and maintain an adequate system of internal accounting controls for international operations. Compliance with the FCPA is expensive and difficult, particularly in countries in which corruption is a recognized problem. In addition, the FCPA presents particular challenges in the pharmaceutical industry, because, in many countries, hospitals are operated by the government, and doctors and other hospital employees are considered foreign officials. Certain payments to hospitals in connection with clinical trials and other work have been deemed to be improper payments to government officials and have led to FCPA enforcement actions.
Many countries, including the United States, also have various lobbying laws and regulations governing the conduct of individuals and companies who interact with government officials. These laws and regulations typically include certain restrictions and disclosure obligations. We believe we are currently in compliance with such laws and regulations. If we, our employees,
or third parties acting on our behalf do not comply with these laws and regulations, we may be subject to civil and criminal penalties.
Many countries, including the United States, restrict the export or import of products to or from certain countries through, for example, bans, sanction programs, and boycotts. Such restrictions may preclude us from supplying products in certain countries, which could limit our growth potential. Furthermore, if we, or third parties acting on our behalf, do not comply with these restrictions, we may be subject to civil and criminal penalties.
Various laws, regulations and executive orders also restrict the use and dissemination outside of the United States, or the sharing with certain non-U.S. nationals, of information classified for national security purposes, as well as certain products and technical data relating to those products. If we continue to expand our presence outside of the United States, it will require us to dedicate additional resources to comply with these laws, and these laws may preclude us from developing, manufacturing, or selling certain products and product candidates outside of the United States, which could limit our growth potential and increase our development costs.
The failure to comply with laws governing international business practices may result in substantial civil and criminal penalties and suspension or debarment from government contracting. The SEC also may suspend or bar issuers from trading securities on U.S. exchanges for violations of the FCPA’s accounting provisions.
MANUFACTURING RISKS
Disruption at, damage to or destruction of our manufacturing facilities could impede our ability to manufacture anthrax vaccines, ACAM2000 or our other products, as well as deliver our CDMO services, which would harm our business, financial condition, operating results and cash flows.
Any further interruptions in our manufacturing operations could result in our inability to produce our products and product candidates for delivery to satisfy the demands of our customers in a timely manner, which would reduce our revenues and materially harm our business, financial condition, operating results and cash flows. A number of factors could cause interruptions, including:
•equipment malfunctions or failures;
•technology malfunctions;
•cyber-attacks;
•work stoppages or slowdowns, particularly due to the impact of COVID-19;
55
EMERGENT BIOSOLUTIONS INC.
•civil unrest and protests, including by animal rights activists;
•injunctions;
•damage to or destruction of one or more facilities;
•FDA facility inspection findings/recommendations; and
•product contamination or tampering.
Providers of PHT countermeasures could be subject to an increased risk of terrorist activities. The USG has designated both our Lansing, Michigan and our Bayview bulk manufacturing facility in Baltimore, Maryland as facilities requiring additional security. Although we continually evaluate and update security measures, there can be no assurance that any additional security measures would protect these facilities from terrorist efforts determined to disrupt our manufacturing activities.
The factors listed above could also cause disruptions at our other facilities. We do not have any redundant manufacturing facilities for any of our marketed products. Accordingly, any damage to, or disruption or destruction of one or more of our facilities could impede our ability to manufacture our marketed products, our product candidates and our ability to produce products for external customers, result in losses and delays, including delays in the performance of our contractual obligations or delays in our clinical trials, any of which could be costly to us and materially harm our business, financial condition, operating results and cash flows.
Problems may arise during the production of our marketed products and product candidates, as well as those we produce for our CDMO customers, due to the complexity of the processes involved in their manufacturing and shipment. Significant delays in product manufacturing or development and our ability to ramp up production to meet the needs of our customers could cause delays in recognizing revenues, which would harm our business, financial condition, operating results and cash flows.
The majority of our products and product candidates are biologics. Manufacturing biologics, especially in large quantities, is complex. The products must be made consistently and in compliance with a clearly defined manufacturing process. Problems during manufacturing may arise for a variety of reasons, including problems with raw materials, equipment malfunction and failure to follow specific protocols and procedures. Slight deviations anywhere in the manufacturing process, including obtaining materials, maintaining master seed or cell banks and preventing genetic drift, seed or cell growth, fermentation,
contamination including from particulates among other things, filtration, filling, labeling, packaging, storage and shipping, potency and stability issues and other quality control testing, may result in lot failures or manufacturing shut-downs, delays in the release of lots, product recalls, spoilage or regulatory action. Such deviations may require us to revise manufacturing processes or change manufacturers. Additionally, as our equipment ages, it will need to be replaced, which has the potential to result in similar consequences. Success rates can also vary dramatically at different stages of the manufacturing process, which can reduce yields and increase costs. From time to time, we may experience deviations in the manufacturing process that may take significant time and resources to resolve and, if unresolved, may affect manufacturing output and could cause us to fail to satisfy customer orders or contractual commitments, lead to a termination of one or more of our contracts, lead to delays in our clinical trials, result in litigation or regulatory action against us, including the issuance of Forms FDA 483. warning letters and other restrictions on the marketing or manufacturing of a product, or cause the FDA to cease releasing product until the deviations are explained and corrected, any of which could be costly to us, damage our reputation and negatively impact our business. For example in April 2021, we temporarily stopped manufacturing bulk drug substance material for Johnson & Johnson’s COVID-19 vaccine at our Baltimore Bayview facility after issues were identified in a viral vaccine drug substance batch.
Additionally, if changes are made to the manufacturing process, we may be required to provide the FDA with pre-clinical and clinical data showing the comparable identity, strength, quality, purity or potency of any impacted products before and after the changes.
We are contractually required to ship our biologic products at a prescribed temperature range and variations from that temperature range could result in loss of product and could significantly and adversely impact our revenues, which would harm our business, financial condition, operating results and cash flows.
In addition, we may not be able to ramp up our manufacturing processes to meet the rapidly changing demand or specifications of our customers on the desired timeframe, if at all. For example, we have not previously had to ramp our organization for a commercial launch of any product at the current pace required to address treatments related to COVID-19 and doing so in a pandemic environment with an urgent, critical global need creates unique manufacturing challenges, challenges related to distribution channels, and the need to establish teams of people with the relevant skills. Our inability to ramp up manufacturing to meet the demand or specifications
56
EMERGENT BIOSOLUTIONS INC.
of our customers or the inability to timely obtain regulatory authorization to produce the products or product candidates of our customers could also harm our business, financial condition, operating results and cash flows.
Our products and product candidates procured by the USG and other customers require us to perform tests for and meet certain potency and lot release standards prescribed by the FDA and other agencies, which may not be met on a timely basis or at all.
Our products and product candidates procured by the USG and other customers require us to perform tests for and meet certain potency and lot release standards prescribed by the FDA and other agencies, which may not be met on a timely basis or at all. We are unable to sell any products and product candidates that fail to satisfy such testing specifications. For example, we must provide the FDA with the results of certain tests, including potency tests, before certain lots are released for sale. Potency testing of each applicable lot is performed against qualified control lots that we maintain. We continually monitor the status of such reference lots for FDA compliance and periodically produce and qualify a new reference lot to replace the existing reference lot. If we are unable to satisfy USG requirements for the release of our products or product candidates, our ability to supply such products and product candidates to authorized buyers would be impaired until such time as we become able to meet such requirements, which could materially harm our future business, financial condition, operating results and cash flows.
Our operations, including our use of hazardous materials, chemicals, bacteria and viruses, require us to comply with regulatory requirements and expose us to significant potential liabilities.
Our operations involve the use of hazardous materials, including chemicals, bacteria and viruses, and may produce dangerous waste products. Accordingly, we, along with the third parties that conduct clinical trials and manufacture our products and product candidates on our behalf, are subject to federal, state, local and foreign laws and regulations that govern the use, manufacture, distribution, storage, handling, exposure, disposal and recordkeeping with respect to these materials. Under the Federal Select Agent Program, pursuant to the Public Health Security and Bioterrorism Preparedness and Response Act, we are required to register with and be inspected by the CDC and the Animal and Plant Health Inspection Service if we have in our possession, or if we use or transfer, select biological agents or toxins that could pose a threat to public health and safety, to animal or plant health or to animal or plant products. This legislation requires stringent safeguards and security measures for these select agents and toxins, including
controlled access and the screening of entities and personnel and establishes a comprehensive national database of registered entities. We are also subject to a variety of environmental and occupational health and safety laws. Compliance with current or future laws and regulations in this area can require significant costs and we could be subject to substantial fines and penalties in the event of noncompliance. In addition, the risk of contamination or injury from these materials cannot be completely eliminated. In such event, we could be held liable for substantial civil damages or costs associated with the cleanup of hazardous materials. From time to time, we have been involved in remediation activities and may be so involved in the future. Any related cost or liability might not be fully covered by insurance, could exceed our resources and could have a material adverse effect on our business, financial condition, operating results and cash flows. In addition to complying with environmental and occupational health and safety laws, we must comply with special regulations relating to biosafety administered by the CDC, HHS, U.S. Department of Agriculture and the DoD, as well as regulatory authorities in Canada.
COMPETITIVE AND POLITICAL RISKS
Development and commercialization of pharmaceutical products, including for PHT preparedness, are routinely subject to evolving private and public sector competition.
The development and commercialization of new biopharmaceutical and medical technology products is highly competitive and subject to rapid technological advances. We may face future competition from other companies and governments, universities and other non-profit research organizations in respect to our products, any products that we acquire, our current product candidates and any products we may seek to develop or commercialize in the future. The market for current products can be subject to development of safer, more effective, more convenient or less costly products. The market for current products can also depend on what resources can be devoted to marketing or selling products, or how companies are positioned to adapt more quickly to new technologies, respond to scientific advances or patient preferences and needs, initiate or withstand substantial price competition and/or procure third-party licensing and collaborative arrangements.
There are a number of companies with products or product candidates addressing PHT preparedness that are competing with us for both USG procurement and development resources. Factors to consider include competitors' financial, technical and marketing resources as well as potential leverage that their intellectual property estates may offer.
57
EMERGENT BIOSOLUTIONS INC.
Any reduction in demand for our products or reduction or loss of development funding for our products or product candidates in favor of a competing product could lead to a loss of market share for our products and cause reduced revenues, margins and levels of profitability for us, which could adversely affect our business, financial condition, operating results and cash flows.
Our Biologic Products may face risks of competition from biosimilar manufacturers.
Biological products and product candidates, otherwise referred to as our “Biologic Products,” can be affected by the approval and entry of “biosimilars” in the United States and other jurisdictions. Biosimilar drugs are “highly similar,” but close enough in duplication to accomplish the same therapeutic and clinical result. Biologic Products in our current pipeline include AV7909, BioThrax, and ACAM2000. If a biosimilar version of one of our Biologic Products were approved, it could have a material adverse effect on the sales and gross profits of the affected Biologic Product and could adversely affect our business, financial condition, operating results and cash flows.
NARCAN® Nasal Spray may be subject to potential competition.
Although NARCAN® Nasal Spray is the first FDA-approved needle-free naloxone nasal spray for the emergency reversal of opioid overdoses and has advantages over certain other treatments, we expect the treatment to face additional competition. For example, on April 30, 2021, the FDA approved Kloxxado, a branded product developed by Hikma Pharmaceuticals, Inc. which delivers a higher dose naloxone nasal spray. In addition, Orexo AB and Harm Reduction Therapeutics both have development programs for novel naloxone nasal spray formulations intended for use in opioid overdose reversal.
NARCAN® Nasal Spray faces additional branded competition from other injectable naloxone, auto-injectors and improvised nasal kits including Amphastar Pharmaceuticals, Inc.'s naloxone injection product and Kaléo's EVZIO™ (naloxone HCI injection) Auto-Injector. NARCAN® Nasal Spray may face additional branded competition in the future.
With respect to potential generic competition, Teva Pharmaceuticals Industries Limited and Teva Pharmaceuticals USA (collectively, Teva) (in 2016) and Perrigo UK FINCO Limited Partnership (Perrigo) (in 2018) each filed an ANDA seeking regulatory approval to market a generic version of NARCAN® Nasal Spray. The outcome of ANDA litigation with Teva is pending following the August 2, 2021 hearing at the Court of Appeals for the Federal Circuit, following our appeal of the June 5, 2020 decision of the U.S. District Court for the District of New Jersey. A current at-risk launch by
Teva remains possible. Settlement with Perrigo regarding their ANDA filing was entered on February 12, 2020 providing for a license effective January 5, 2033, or earlier under certain circumstances, including those related to the outcome of the current Teva litigation or future ANDA filers.
Sales of generic versions of NARCAN® Nasal Spray at prices lower than our branded product have the potential to erode our sales and could impact our product revenue related to NARCAN® Nasal Spray. For example, certain U.S. state laws allow for, and in some instances in the absence of specific instructions from the prescribing physician, mandate the dispensing of generic products rather than branded products where a generic version is available. In addition, in January 2019, the FDA released new proposed template Drug Facts Labels to assist sponsors of investigational naloxone nasal sprays and auto-injectors seeking approval from the FDA for over-the-counter naloxone products.
Political or social factors may delay or impair our ability to market our products and may require us to spend significant management time and financial resources to address these issues.
Products developed to counter the potential impact of PHTs are subject to changing political and social environments. The political responses and social awareness of the risks of these threats on military personnel or civilians and the level of emphasis placed on such risks by the USG may vary over time. If the threat of terrorism were to decline, then the public perception of the risk on public health and safety may be reduced. This perception, as well as political or social pressures, could delay or cause resistance to bringing our products in development to market or limit pricing or purchases of our products, any of which could negatively affect our revenues and our business, financial condition, operating results and cash flows.
In addition, substantial delays or cancellations of purchases could result from protests or challenges from third parties. Lawsuits brought against us by third parties or activists, even if not successful, could require us to spend significant management time and financial resources defending the related litigation and could potentially damage the public's perception of us and our products. Any publicity campaigns or other negative publicity may adversely affect the degree of market acceptance of our PHT countermeasures and thereby limit the demand for our products, which would adversely affect our business, financial condition, operating results and cash flows.
58
EMERGENT BIOSOLUTIONS INC.
INTELLECTUAL PROPERTY RISKS
Protection of our intellectual property rights is an important tool for sustaining our business and the failure to do so could impact our financial condition, operating results, and cash flows.
We actively seek to protect intellectual property rights related to our Company's assets, including patent rights, trademark rights, trade secrets and proprietary confidential information, through defense and enforcement of existing rights and pursuit of protection on new and arising innovations.
Obtaining, maintaining and defending our intellectual property rights in the United States and other countries remains a critical component of the development and commercialization of our Company's assets.
Some of the risks associated with procurement, maintenance and enforcement of intellectual property rights include changes in patent laws or administrative patent office rules, evolving criteria and eligibility of obtaining patent protection on particular subject matter, the validity and enforceability of our intellectual property rights, the potential scope of coverage of our intellectual property rights, and/or the availability or strength of legal remedies in a particular country to defend and enforce intellectual property rights.
Other risks include associated costs, such as costs of patent prosecution and maintenance and costs associated with post-grant challenges. For example, such costs include inter partes review (IPR) proceedings in the United States and oppositions in Europe, as well as costs associated with litigating and enforcing patent and trademark rights, such as the costs associated with appealing the decision related to the pending patent litigation involving NARCAN® Nasal Spray discussed above.
Additional risks include limitations on our extent or ability to procure, maintain or defend intellectual property rights associated with in-licensed or acquired intellectual property, where, for example, third parties may have the first right to maintain or defend intellectual property rights in which we have an interest, or may pursue strategies that are divergent to the interest of our Company.
Third party challenges for patent infringement could impact our business, financial condition, operating results, and cash flows.
Challenges by third parties for alleged patent infringement could delay or affect the development and commercialization of our products. Such challenges, while ongoing, could be costly, requiring and utilizing company resources. Such challenges, if successful, may impact marketing or launch of products, or require ongoing license and/or royalty fees associated with
potential settlement agreements. These may have the potential to materially harm our business, financial condition, operating results, and cash flows.
Intellectual property licenses with third parties carry risks of challenges, which may be costly and time consuming and could impact the commercialization of our products.
We are a party to a number of license agreements and expect to enter into additional license agreements in the future. Such license agreements or collaboration arrangements can be subject to challenges if interests or expectations under such license agreements diverge. Such challenges may be costly, risk time and resources, and could delay or impact development, commercialization or launch of our products.
Potential loss of proprietary information and know-how generally carries the risk of reducing the value of our technology and products.
We also rely upon unpatented proprietary technology, processes, and know-how, particularly as to our proprietary manufacturing processes. These types of confidential information and trade secrets can be difficult to protect. We seek to protect this confidential information, in part, through agreements with our employees, consultants, and third parties, as well as confidentiality policies and audits, although these may not always be successful in protecting our trade secrets and confidential information.
One or more of our products could be subject to early competition from generic drugs and biosimilars.
One or more of our products is approved as a drug product under the provisions of the FDCA, which may render it susceptible to potential competition from generic manufacturers via the Hatch-Waxman Act and ANDA process. Other of our products may be susceptible to challenges by entry of biosimilars through the route established under the Biologics Price Competition and Innovation Action of 2009.
Although we intend to vigorously enforce our intellectual property rights, there can be no assurance that we will prevail in our enforcement or defense of our patent rights. Our existing patents could be invalidated, found unenforceable, or found not to cover a generic form of our product.
RISKS RELATED TO RELIANCE ON THIRD PARTIES
The loss of any of our non-exclusive, sole-source or single source suppliers, a shortage of related supplies or an increase in the price of inventory supplied to us could have an adverse effect on our business, financial condition and results of operations.
We purchase certain supplies used in our manufacturing processes from non-exclusive, or single
59
EMERGENT BIOSOLUTIONS INC.
sources due to quality considerations, costs or constraints resulting from regulatory requirements. We depend on certain single-source suppliers for key materials and services necessary to manufacture the majority of our products and certain product candidates. For example, we rely on a single-source supplier to provide us with Alhydrogel in sufficient quantities to meet our needs to manufacture AV7909 and BioThrax and the specialty plasma in our hyperimmune specialty plasma products and certain ingredients for ACAM2000. We also rely on single-source suppliers for the materials necessary to produce NARCAN®Nasal Spray, such as the naloxone active pharmaceutical ingredient and other excipients, along with the vial, stopper and device.
Where a particular single-source supply relationship is terminated, we may not be able to establish additional or replacement suppliers for certain components or materials quickly. This is largely due to the FDA approval system, which mandates validation of materials prior to use in our products, and the complex nature of manufacturing processes. In addition, we may lose a sole-source supplier due to, among other things, the impact of COVID-19 on such supplier, the acquisition of a supplier by a competitor (which may cause the supplier to stop selling its products to us) or the bankruptcy of such a supplier, which may cause the supplier to cease operations. Any reduction or interruption by a sole-source supplier of the supply of materials or key components used in the manufacturing of our products or product candidates, a reduction in quality or an increase in the price of those materials or components could adversely affect us. If we are unable to locate or establish alternative suppliers, our ability to manufacture our products and product candidates could be adversely affected and could harm our revenues, cause us to fail to satisfy contractual commitments, lead to a termination of one or more of our contracts or lead to delays in our clinical trials, any of which could be costly to us and otherwise materially harm our business, financial condition, operating results and cash flows.
We depend on third parties to conduct many of our clinical and non-clinical trials. If these third parties do not perform as contractually required or as we expect, we may not be able to obtain regulatory approval for or commercialize our product candidates and, as a result, our business, financial condition, operating results and cash flows may suffer.
We depend on third parties, such as independent clinical investigators, contract research organizations and other third-party service providers to conduct the clinical and non-clinical trials of our product candidates and expect to continue to do so. We rely heavily on these third parties for successful execution of our clinical and non-clinical trials, but do not exercise day-to-day control over their activities. Our reliance on
these service providers does not relieve us of our regulatory responsibilities, including ensuring that our trials are conducted in accordance with good clinical practice regulations and the plan and protocols contained in the relevant regulatory application. In addition, these organizations may not complete these activities on our anticipated or desired timeframe. We also may experience unexpected cost increases that are beyond our control. Problems with the timeliness or quality of the work of a contract research organization may lead us to seek to terminate the relationship and use an alternative service provider, which may prove difficult, costly and result in a delay of our trials. Any delay in or inability to complete our trials could delay or prevent the development, approval and commercialization of our product candidates.
In certain cases, government entities and non-government organizations conduct studies of our product candidates, and we may seek to rely on these studies in applying for marketing approval for certain of our product candidates. These government entities and non-government organizations have no obligation or commitment to us to conduct or complete any of these studies or clinical trials and may choose to discontinue these development efforts at any time. Furthermore, government entities depend on annual Congressional appropriations to fund their development efforts, which may not be approved.
If we are unable to obtain any necessary third-party services on acceptable terms or if these service providers do not successfully carry out their contractual duties or meet expected deadlines, our efforts to obtain regulatory approvals for our product candidates may be delayed or prevented.
LEGAL AND REPUTATIONAL RISKS
Pending litigation and legal proceedings and the impact of any finding of liability or damages could adversely impact our business, financial condition and results of operations.
We are currently involved in numerous lawsuits, including stockholder derivative and putative class action lawsuits and anticipate that we will continue to be a target of such lawsuits in the future due to the volatility of our stock price. Certain of these actions include, and future actual or threatened legal actions may include, claims for substantial and indeterminate amounts of damages, or may result in other actions adverse to us.
For example, multiple purported class action lawsuits have been filed against us and certain of our current and former senior officers in the United States District Court for the District of Maryland seeking unspecified damages on behalf of a putative class of persons who purchased or otherwise acquired shares of our common stock during various date ranges. The
60
EMERGENT BIOSOLUTIONS INC.
complaints, allege, among other things, that we made materially false and misleading statements regarding our procedures and quality controls relating to vaccine production, in violation of federal securities laws. As another example, a purported stockholder derivative lawsuit was filed in the United States District Court for the District of Maryland on behalf of the Company against certain of our current and former officers and directors for breach of fiduciary duties, waste of corporate assets, and unjust enrichment. In addition to monetary damages, the complaint seeks the implementation of multiple corporate governance and internal policy changes.
The results of these lawsuits and possible other future legal proceedings cannot be predicted with certainty. Accordingly, we cannot determine whether our insurance coverage would be sufficient to cover related costs or potential losses, if any. Regardless of merit, litigation can be both time-consuming and disruptive to our operations and cause significant expense and diversion of management’s attention. If we do not prevail in the purported class action lawsuits or in other future legal proceedings, we may be faced with significant monetary damages or injunctive relief against us that may adversely affect our business, financial condition and results of operations.
We rely significantly on information technology systems and any failure, inadequacy, interruption or security lapse of that technology, including any cyber security incidents, could harm our ability to operate our business effectively or result in data leakage of proprietary and confidential business and employee information.
Our business is increasingly dependent on critical, complex and interdependent information technology systems, including Internet-based systems, to support business processes as well as internal and external communications. We also have contracted with the USG and pharmaceutical companies, such as Johnson & Johnson, for the development and manufacture of a significant quantity of COVID-19 vaccines, and separately we are working on a proprietary COVID-19 therapeutic with support from the USG and other private sector entities, which has raised our security profile, and heightened potential risks that malicious actors may seek to disrupt our systems or misappropriate our information. The size and complexity of our computer systems make them potentially vulnerable to interruption, invasion, computer viruses, destruction, malicious intrusion and additional related disruptions, which may result in the impairment of production and key business processes. Our systems are also potentially vulnerable to data security breaches through employee error, phishing scams and malfeasance, which may expose sensitive data to unauthorized persons. No system of protection
is adequate to protect against all such threats, even if they are deemed to be industry standard, and there can be no assurance that we will be able to repel any such attacks. Data security breaches could lead to the loss of trade secrets or other intellectual property or the public exposure of personal information, including sensitive personal information, of our employees, clinical trial patients, customers and others. Responding to any such threats may also be expensive and time-consuming.
A significant business disruption or a breach in security resulting in misappropriation, theft or sabotage with respect to proprietary and confidential business and employee information could result in significant financial losses, legal, business or reputational harm to us, compromise our business prospects and our commitments to the USG or other customers, any of which could materially and adversely affect our business, financial condition and operating results.
Our work on public health threats has exposed us to criticism and may expose us to further criticism, from the media, government personnel, and others, that can negatively effect our share price, reputation, operations, and our ability to attract and retain talent.
Our work on public health threats, including manufacturing issues at our Baltimore Bayview facility, has exposed us to criticism and may expose us to additional potential criticism, from the media, government personnel, and others. In addition, our work on public health threats has exposed us to governmental inquiries and investigations, including by Congress and other government agencies. For example, a joint panel of the U.S. House of Representatives launched an investigation into, among other things, the cause of the previously mentioned cross-contamination issues identified in a viral vaccine drug substance batch at the Baltimore Bayview facility. Such criticism can be particularly acute during a public health emergency like the COVID-19 pandemic. The unfavorable media coverage and increased government scrutiny, including the Congressional inquiry, could further harm our reputation, distract management’s attention from our operations, and impact our ability to attract and retain talent and result in further declines to our share price. We have already incurred significant legal costs to respond to government inquiries and are likely to incur additional costs. Any adverse actions by government authorities may result in significant civil or criminal fines or penalties, all of which could adversely impact our financial condition, operating results and cash flows.
We face product liability exposure, which could cause us to incur substantial liabilities and negatively
61
EMERGENT BIOSOLUTIONS INC.
affect our business, financial condition and results of operations.
We face an inherent risk of product liability exposure related to the sale of our products, any other products that we successfully acquire or develop and the testing of our product candidates in clinical trials.
One measure of protection against such lawsuits is coverage under the PREP Act, which was signed into law in December 2005. The PREP Act creates liability protection for manufacturers of biodefense countermeasures when the Secretary of HHS issues a declaration for their manufacture, administration or use. A PREP Act declaration is meant to provide liability protection from all claims under federal or state law for loss arising out of the administration or use of a covered countermeasure under a government contract. The Secretary of HHS has issued PREP Act declarations identifying certain of our products, namely BioThrax, ACAM2000, raxibacumab, Anthrasil, BAT and VIGIV, as covered countermeasures. These declarations expire in 2022. Manufacturers are not entitled to protection under the PREP Act in cases of willful misconduct or for cases brought in non-U.S. tribunals or under non-U.S. law. We cannot predict whether the Secretary of HHS will renew the declarations when they expire, whether Congress will fund the relevant PREP Act compensation programs, or whether the necessary prerequisites for immunity would be triggered with respect to our products or product candidates.
Additionally, certain of our products, namely BioThrax and RSDL, are certified anti-terrorism products covered under the protections of the SAFETY Act. The SAFETY Act creates product liability limitations for qualifying anti-terrorism technologies for claims arising from or related to an act of terrorism. Although we are entitled to the benefits of the SAFETY Act for BioThrax and RSDL, the SAFETY Act may not provide adequate protection from claims made against us.
If we cannot successfully defend ourselves against future claims that our products or product candidates caused injuries and if we are not entitled to indemnity by the USG, or the USG does not honor its obligations to us under the PREP Act or SAFETY Act, or if the liability protections under the PREP Act and SAFETY Act are not adequate to cover all claims, we may incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
•decreased demand or withdrawal of a product;
•injury to our reputation;
•withdrawal of clinical trial participants;
•costs to defend the related litigation;
•substantial monetary awards to trial participants or patients;
•loss of revenue; and
•an inability to commercialize products that we may develop.
The amount of insurance that we currently hold may not be adequate to cover all liabilities that we may incur. Further product liability insurance may be difficult and expensive to obtain. We may not be able to maintain insurance coverage at a reasonable cost and we may not be able to obtain insurance coverage that will be adequate to satisfy all potential liabilities. For example, we may not have sufficient insurance against potential liabilities associated with a possible large-scale deployment of BioThrax as a countermeasure to a bioterrorism threat. We rely on PREP Act protection for BioThrax, raxibacumab, ACAM2000, Anthrasil, BAT and VIGIV, and SAFETY Act protection for BioThrax and RSDL in addition to our insurance coverage to help mitigate our product liability exposure for these products. Additionally, potential product liability claims related to our commercial products, including NARCAN® Nasal Spray, Vivotif and Vaxchora, may be made by patients, health care providers or others who sell or consume these products. Such claims may be made even with respect to those products that possess regulatory approval for commercial sale. Claims or losses in excess of our product liability insurance coverage could have a material adverse effect on our business, financial condition, operating results and cash flows.
FINANCIAL RISKS
We have incurred significant indebtedness in connection with our acquisitions and servicing our debt requires a significant amount of cash. We may not have sufficient cash flow from our operations to pay our substantial debt.
Our ability to make scheduled payments of the principal of, to pay interest on or to further refinance our indebtedness depends on our future performance, which is subject to economic, financial, competitive and other factors beyond our control. We may also seek additional debt financing to support our ongoing activities or to provide additional financial flexibility. Debt financing can have significant adverse consequences for our business, including:
•requiring us to dedicate a substantial portion of cash flows from operations to payment on our debt, which would reduce available funds for other corporate initiatives;
•increasing the amount of interest that we have to pay on debt with variable interest rates, if
62
EMERGENT BIOSOLUTIONS INC.
market rates of interest increase, to the extent we are unable to offset such risk through our hedging instruments;
•subjecting us, as under our Senior Secured Credit Facilities and the indenture governing the 3.875% Senior Unsecured Notes due 2028 (Senior Unsecured Notes), to restrictive covenants that reduce our ability to take certain corporate actions, acquire companies, products or technology, or obtain further debt financing;
•requiring us to pledge our assets as collateral, which could limit our ability to obtain additional debt financing;
•limiting our flexibility in planning for, or reacting to, general adverse economic and industry conditions; and
•placing us at a competitive disadvantage compared to our competitors that have less debt, better debt servicing options or stronger debt servicing capacity.
We may not have sufficient funds or be able to obtain additional financing to pay the amounts due under our indebtedness. In addition, failure to comply with the covenants under our Senior Secured Credit Facilities and other debt agreements, including the maintenance of a specified consolidated net leverage ratio and debt service coverage ratio under our Senior Secured Credit Facilities, could result in an event of default under those agreements. An event of default could result in the acceleration of amounts due under a particular debt agreement and a cross default and acceleration under other debt agreements, and we may not have sufficient funds to pay or be able to obtain additional financing to make any accelerated payments. Under these circumstances, our lenders could seek to enforce security interests in our assets securing our indebtedness.
Our current indebtedness restricts and any additional debt financing may restrict the operation of our business and limit the cash available for investment in our business operations.
The Senior Secured Credit Facilities include a $450 million Term Loan Facility and the ability to borrow up to $600 million under our Revolving Credit Facility, of which we had outstanding borrowings of approximately $405.0 million and no outstanding balance, respectively, as of September 30, 2021. On August 7, 2020, we completed an offering of $450 million aggregate principal amount of Senior Unsecured Notes, of which $353 million of the net proceeds were used to pay down our Revolving Credit Facility. We may also seek additional debt financing to support our ongoing activities or to provide additional financial
flexibility. Debt financing can have significant adverse consequences for our business, including:
•the level, timing and cost of product sales and CDMO services;
•the extent to which we acquire or invest in and integrate companies, businesses, products or technologies;
•the acquisition of new facilities and capital improvements to new or existing facilities;
•the payment obligations under our indebtedness;
•the scope, progress, results and costs of our development activities;
•our ability to obtain funding from collaborative partners, government entities and non-governmental organizations for our development programs;
•the extent to which we repurchase common stock under any future share repurchase program; and
•the costs of commercialization activities, including product marketing, sales and distribution.
Our hedging program is subject to counterparty default risk.
We manage our interest rate risk in part by entering into interest rate swaps with a number of counterparties to swap a portion of our indebtedness that is based on variable interest rates to a fixed rate. As a result, we are subject to the risk that the counterparty to one or more of these contracts defaults on its performance under the contract. During an economic downturn, the counterparty's financial condition may deteriorate rapidly and with little notice and we may be unable to take action to protect our exposure. In the event of a counterparty default, we could incur losses, which may harm our business and financial condition. In the event that one or more of our counterparties becomes insolvent or files for bankruptcy, our ability to eventually recover any losses suffered as a result of that counterparty's default may be limited by the liquidity of the counterparty.
We may require significant additional funding and be unable to raise capital when needed or on acceptable terms, which would harm our ability to grow our business, and our results of operations and financial condition.
If our capital resources are insufficient to meet our future capital requirements, we will need to finance our
63
EMERGENT BIOSOLUTIONS INC.
cash needs through public or private equity or debt offerings, bank loans or collaboration and licensing arrangements. In August 2021, we filed an automatic shelf registration statement, which immediately became effective under SEC rules. For so long as we continue to satisfy the requirements to be deemed a “well-known seasoned issuer” under SEC rules (which include, among other things, the timely filing of our reports under the Exchange Act and maintenance of at least $700 million of public float or issuing an aggregate amount of $1 billion of non-convertible securities, other than common stock, in registered offerings for cash during the past three years), this shelf registration statement, effective until August 9, 2024, allows us to issue an unrestricted amount of equity, debt and certain other types of securities through one or more future primary or secondary offerings. If we do not file a new shelf registration statement prior to August 9, 2024, the existing shelf registration statement will expire, and we will not be able to publicly raise capital or issue debt until a new registration statement is filed and becomes effective. There can be no assurance that we will be eligible to file an automatically effective shelf registration statement at a future date when we may need to raise funds publicly.
If we raise funds by issuing equity securities, our stockholders may experience dilution. Debt financing, if available, may involve agreements that include covenants, like those contained in our Senior Secured Credit Facilities and the indenture governing the Senior Unsecured Notes, limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures, pursuing acquisition opportunities or declaring dividends. If we raise funds through collaboration and licensing arrangements with third parties, it may be necessary to relinquish valuable rights to our technologies or product candidates or grant licenses on terms that may not be favorable to us. Our Senior Secured Credit Facilities as well as the indenture governing the Senior Unsecured Notes restrict our ability to incur additional indebtedness.
Economic conditions may make it difficult to obtain financing on attractive terms, or at all. If financing is unavailable or lost, our business, operating results, financial condition and cash flows would be adversely affected, and we could be forced to delay, reduce the scope of or eliminate many of our planned activities.
We may not maintain profitability in future periods or on a consistent basis.
Although we have been profitable on an annual basis since becoming a public company, we have not been profitable for every quarter during that time. Our profitability has been substantially dependent on product sales, which historically have fluctuated significantly from quarter to quarter, and we expect that
they will continue to fluctuate significantly based primarily on the timing of our fulfillment of orders from the USG. We may not be able to achieve consistent profitability on a quarterly basis or sustain or increase profitability on an annual basis.
The expansion of our international operations increases our risk of exposure to credit losses.
As we continue to expand our business activities with foreign governments in certain countries that have experienced deterioration in credit and economic conditions or otherwise, our exposure to uncollectible accounts will rise. Global economic conditions and liquidity issues in certain countries have resulted and may continue to result in delays in the collection of accounts receivable and may result in credit losses. Future governmental actions and customer specific actions may require us to re-evaluate the collectability of our accounts receivable and we may potentially incur credit losses that materially impact our operating results.
A substantial portion of our indebtedness bears interest at variable interest rates based on LIBOR and certain of our financial contracts are also indexed to LIBOR. Changes in the method of determining LIBOR, or the replacement of LIBOR with an alternative reference rate, may adversely affect interest rates on our current or future indebtedness and may otherwise adversely affect our financial condition and results of operations.
In July 2017, the Financial Conduct Authority, the authority that regulates the London Inter-bank Offered Rate (LIBOR) announced that it intended to stop compelling banks to submit rates for the calculation of LIBOR.
On November 30, 2020, the International Exchange (ICE) Benchmark Association, which administrates LIBOR, announced that it intends to begin a phase out of LIBOR at the end of 2021, by ceasing (i) entering into new contracts that use LIBOR as a reference rate by December 31, 2021 and (ii) publication of two LIBOR rates (one-week and two-month) after December 31, 2021, while the remaining LIBOR rates (overnight, one-month, three-month, six-month and 12-month) will be retired on June 30, 2023. It is unclear if LIBOR will cease to exist at that time or if new methods of calculating LIBOR will be established such that it continues to exist after 2023. We have certain financial contracts, including the amended credit agreement related to our Senior Secured Credit Facilities and our interest rate swaps, that are indexed to LIBOR. Changes in the method of determining LIBOR, or the replacement of LIBOR with an alternative reference rate, may adversely affect interest rates on our current or future indebtedness. Any transition process may involve, among other things, increased volatility or illiquidity in markets for instruments that rely on LIBOR, reductions in the value of certain
64
EMERGENT BIOSOLUTIONS INC.
instruments or the effectiveness of related transactions such as hedges, increased borrowing costs, uncertainty under applicable documentation, or difficult and costly consent processes. The transition away from LIBOR may result in increased expenses, may impair our ability to refinance our indebtedness or hedge our exposure to floating rate instruments, or may result in difficulties, complications or delays in connection with future financing efforts, any of which could adversely affect our financial condition and results of operations.
RISKS RELATED TO STRATEGIC ACQUISITIONS AND COLLABORATIONS
Our strategy of generating growth through acquisitions may not be successful.
Our business strategy includes growing our business through acquisition and in-licensing transactions. We may not be successful in identifying, effectively evaluating, structuring, acquiring or in-licensing, and developing and commercializing additional products on favorable terms, or at all. Competition for attractive product opportunities is intense and may require us to devote substantial resources, both managerial and financial, to an acquisition opportunity. A number of more established companies are also pursuing strategies to acquire or in-license products in the biopharmaceutical field. These companies may have a competitive advantage over us due to their size, cash resources, cost of capital, effective tax rate and greater clinical development and commercialization capabilities.
Acquisition efforts can consume significant management attention and require substantial expenditures, which could detract from our other programs. In addition, we may devote significant resources to potential acquisitions that are never completed. Even if we are successful in acquiring a company or product, it may not result in a successfully developed or commercialized product or, even if an acquired product is commercialized, competing products or technologies could render a product noncompetitive, uneconomical or obsolete. Moreover, the cost of acquiring other companies or in-licensing products could be substantial, and in order to acquire companies or new products, we may need to incur substantial debt or issue dilutive securities.
If we are unsuccessful in our efforts to acquire other companies or in-license and develop additional products, or if we acquire or in-license unproductive assets, it could have a material adverse effect on the growth of our business, and we could be compelled to record significant impairment charges to write-down the carrying value of our acquired intangible assets, which could materially harm our business, financial condition, operating results and cash flows.
Our failure to successfully integrate acquired businesses and/or assets into our operations could adversely affect our ability to realize the benefits of such acquisitions and, therefore, to grow our business.
We may not be able to integrate any acquired business successfully or operate any acquired business profitably. In addition, cost synergies, if achieved at all, may be less than we expect, or may take greater time to achieve than we anticipate.
Issues that could delay or prevent successful integration or cost synergies of an acquired business or products include, among others:
•retaining existing customers and attracting new customers;
•retaining key employees;
•diversion of management attention and resources;
•conforming internal controls, policies and procedures, business cultures and compensation programs;
•consolidating corporate and administrative infrastructures;
•successfully executing technology transfers and obtaining required regulatory approvals;
•consolidating sales and marketing operations;
•identifying and eliminating redundant and underperforming operations and assets;
•assumption of known and unknown liabilities;
•coordinating geographically dispersed organizations;
•managing tax costs or inefficiencies associated with integrating operations; and
•risks associated with intellectual property rights related to an acquisition or collaboration.
If we are unable to successfully integrate pending and future acquisitions with our existing businesses, or operate any acquired business profitably, we may not obtain the advantages that the acquisitions were intended to create, which may materially adversely affect the growth of our business, financial condition, operating results and cash flows.
RISKS RELATED TO OWNERSHIP OF OUR COMMON STOCK
Our business or our share price could be negatively affected as a result of the actions of shareholders.
65
EMERGENT BIOSOLUTIONS INC.
In recent years, some shareholders have placed increasing pressure on publicly traded companies in our industry and others to effect changes to corporate governance practices, executive compensation practices, social and environmental practices and to undertake certain corporate actions. This may be true even if they only hold a minority of shares. In addition, some institutional investors are increasingly focused on environmental, social and governance (ESG) factors. These investors may be seeking enhanced ESG disclosures or to implement policies adverse to our business. There can be no assurances that shareholders will not publicly advocate for us to make corporate governance changes or engage in certain corporate actions. Responding to challenges from shareholders, such as proxy contests, media campaigns or other public or private means, could be costly and time consuming and could have an adverse effect on our reputation and divert the attention and resources of management and our board, which could have an adverse effect on our business and operational results. Any such shareholder actions or requests, or the mere public presence of shareholders with a reputation for taking such actions among our shareholder base, could also cause the market price of our common stock to experience periods of significant volatility.
Fuad El-Hibri, executive chairman of our Board of Directors, has significant influence over us through his substantial beneficial ownership of our common stock, including an ability to influence the election of the members of our Board of Directors, or delay or prevent a change of control of us.
Mr. El-Hibri has the ability to significantly influence the election of the members of our Board of Directors due to his substantial beneficial ownership of our common stock. As of September 30, 2021, Mr. El-Hibri was the beneficial owner of approximately 9% of our outstanding common stock. As a result, Mr. El-Hibri could exercise substantial influence over corporate actions requiring board or stockholder approval, including a change of control, or any amendment of our certificate of incorporation or by-laws. The control by Mr. El-Hibri may prevent other stockholders from influencing significant corporate decisions. In addition, Mr. El-Hibri's significant beneficial ownership of our shares could present the potential for a conflict of interest.
Provisions in our certificate of incorporation and by-laws and under Delaware law may discourage acquisition proposals, delay a change in control or prevent transactions that stockholders may consider favorable.
Provisions in our certificate of incorporation and by-laws may discourage, delay or prevent a merger,
acquisition or other changes in control that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. These provisions may also prevent or frustrate attempts by our stockholders to replace or remove our management.
These provisions include:
•the classification of our directors;
•limitations on changing the number of directors then in office;
•limitations on the removal of directors;
•limitations on filling vacancies on the board;
•advance notice requirements for stockholder nominations of candidates for election to the Board of Directors and other proposals;
•the inability of stockholders to act by written consent;
•the inability of stockholders to call special meetings; and
•the ability of our Board of Directors to designate the terms of and issue a new series of preferred stock without stockholder approval.
The affirmative vote of holders of our capital stock representing at least 75% of the voting power of all outstanding stock entitled to vote is required to amend or repeal the above provisions of our certificate of incorporation. The affirmative vote of either a majority of the directors present at a meeting of our Board of Directors or holders of our capital stock representing at least 75% of the voting power of all outstanding stock entitled to vote is required to amend or repeal our by-laws.
In addition, we are subject to Section 203 of the Delaware General Corporation Law (Section 203). In general and subject to certain exceptions, Section 203 prohibits a publicly-held corporation from engaging in a business combination with an interested stockholder, generally a person which, together with its affiliates, owns or within the last three years has owned 15% or more of the corporation's voting stock, for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is approved in a prescribed manner. Accordingly, Section 203 may discourage, delay or prevent a change in control of us.
Our Board of Directors may implement a new stockholder rights plan without stockholder approval, which could prevent a change in control of us in instances in which some stockholders may believe a change in control is in their best interests.
66
EMERGENT BIOSOLUTIONS INC.
Our Board of Directors may implement a stockholder rights plan without stockholder approval. We previously implemented a stockholder rights plan, which expired on November 14, 2016. Under our prior stockholder rights plan, we issued to each of our stockholders one preferred stock purchase right for each outstanding share of our common stock. Each right, when exercisable, would have entitled its holder to purchase from us a unit consisting of one one-thousandth of a share of series A junior participating preferred stock at a purchase price of $150 in cash, subject to adjustments. Our stockholder rights plan was intended to protect stockholders in the event of an unfair or coercive offer to acquire us and to provide our Board of Directors with adequate time to evaluate unsolicited offers.
Our Board of Directors may implement a new stockholder rights plan, which may have anti-takeover effects, potentially preventing a change in control of us in instances in which some stockholders may believe a change in control is in their best interests. This could cause substantial dilution to a person or group that attempts to acquire us on terms that our Board of Directors does not believe are in our best interests or those of our stockholders and may discourage, delay or prevent a merger or acquisition that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares.
Our stock price is volatile, and purchasers of our common stock could incur substantial losses.
Our stock price has been, and is likely to continue to be, volatile. The market price of our common stock could fluctuate significantly for many reasons, including in response to the risks described in this “Risk Factors” section, or for reasons unrelated to our operations, such as reports by industry analysts, investor perceptions or negative announcements by our customers, competitors or suppliers regarding their own performance, as well as industry conditions and general financial, economic and political instability. From November 15, 2006, when our common stock first began trading on the New York Stock Exchange, through October 29, 2021, our common stock has traded as high as $137.61 per share and as low as $4.17 per share. Due to fears associated with COVID-19, the stock market has recently experienced extreme volatility and the market for biopharmaceutical companies has generally experienced extreme volatility that has often been unrelated to the operating performance of particular companies. The market price of our common stock may be influenced by many factors, including, among others:
•contracts, decisions and procurement policies by the USG affecting our anthrax vaccines and our other products and product candidates;
•CDMO contracts related to COVID-19 with collaboration partners;
•the success of competitive products or technologies;
•results of clinical and non-clinical trials of our product candidates;
•announcements of acquisitions, financings or other transactions by us;
•litigation or legal proceedings;
•public concern as to the safety of our products;
•termination or delay of a development program;
•the recruitment or departure of key personnel;
•variations in our product revenue and profitability; and
•the other factors described in this “Risk Factors” section.
Because we currently do not pay dividends, investors will benefit from an investment in our common stock only if it appreciates in value.
We currently do not pay dividends on our common stock. Our Senior Secured Credit Facilities and the indenture governing our Senior Unsecured Notes limit and any future debt agreements that we enter into may limit our ability to pay dividends. As a result, capital appreciation, if any, of our common stock will be the sole source of gain for our stockholders based on current expectations.
Future issuances of our common stock or securities convertible into common stock could result in dilution of our stockholders and could cause our share price to decline.
We expect to continue to opportunistically seek access to additional capital to license or acquire additional products, product candidates or companies to expand our operations or for general corporate purposes. To the extent we raise additional capital by issuing equity securities or securities convertible or exchangeable into common stock, our stockholders may experience substantial dilution. We may sell common stock, and we may sell convertible or exchangeable securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell such common stock, convertible or exchangeable securities or other equity securities in subsequent transactions, existing stockholders may be materially diluted.
GENERAL RISKS
67
EMERGENT BIOSOLUTIONS INC.
We have identified a material weakness in our internal control over financial reporting which could, if not remediated, result in material misstatements in our financial statements.
Our management is responsible for establishing and maintaining adequate internal controls over our financial reporting, as defined in Rule 13a-15(f) and 15d-15(f) under the Securities Exchange Act. As disclosed in Item 4 of Part I of this report, we have identified a material weakness in our internal control over financial reporting related to our technical accounting assessment of the BARDA COVID-19 Development Public Private Partnership and CDMO revenue contracts and related accounting judgments primarily focused on (a) the scoping of lease and non-lease components and (b) the recognition of revenue.
A material weakness is defined as a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis. As a result of this material weakness, our management concluded that our internal control over financial reporting was not effective as of the last day of the period covered by this report. We are actively engaged in implementing a remediation plan designed to address this material weakness. If the remedial measures are insufficient to address the material weakness or if additional material weaknesses or significant deficiencies in the internal controls are discovered or occur in the future, the consolidated financial statements may contain material misstatements and we could be required to restate our financial results. There can be no assurance that we will be successful in making these improvements and in remediating our current material weakness in a timely manner, or at all, and we may not prevent future material weaknesses from occurring.
Our success is dependent on our continued ability to attract, motivate and retain key personnel, and any failure to attract or retain key personnel may negatively affect our business.
Because of the specialized scientific nature of our business, our ability to develop products and to compete with our current and future competitors largely depends upon our ability to attract, retain and motivate highly qualified managerial and key scientific and technical personnel (including quality and manufacturing personnel). If we are unable to retain the services of one or more of the principal members of senior management or other key employees, our ability to implement our business strategy could be materially harmed. We face intense competition for qualified employees from biopharmaceutical companies, research organizations and academic
institutions. Attracting, retaining or replacing these personnel on acceptable terms may be difficult and time-consuming given the high demand in our industry for similar personnel. We believe part of being able to attract, motivate and retain personnel is our ability to offer a competitive compensation package, including equity incentive awards. If we cannot offer a competitive compensation package to attract and retain the qualified personnel necessary for the continued development of our business, we may not be able to maintain our operations or grow our business.
68
EMERGENT BIOSOLUTIONS INC.
ITEM 2. UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS
Recent Sales of Unregistered Securities
Not applicable.
Use of Proceeds
Not applicable.
Purchases of Equity Securities
Not applicable.
ITEM 3. DEFAULTS UPON SENIOR SECURITIES
Not applicable.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
ITEM 5. OTHER INFORMATION
Not applicable.
ITEM 6. EXHIBITS
The exhibits required to be filed by Item 601 of Regulation S-K are listed in the Exhibit Index immediately preceding the exhibits hereto.
69
EMERGENT BIOSOLUTIONS INC.
Exhibit Index
Exhibit Number | Description | ||||
| 10.1#† | |||||
| 10.2#† | |||||
| 10.3#† | |||||
| 10.4#† | |||||
| 10.5#† | |||||
| 10.6#† | |||||
| 10.7#† | |||||
| 31.1 # | |||||
| 31.2 # | |||||
| 32.1 # | |||||
| 32.2 # | |||||
| 101 # | The following financial information related to the Company’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2021, formatted in iXBRL (Inline Extensible Business Reporting Language): (i) the Condensed Consolidated Balance Sheets, (ii) the Condensed Consolidated Statements of Operations, (iii) the Condensed Consolidated Statements of Comprehensive Income, (iv) the Condensed Consolidated Statements of Cash Flows, (v) the Condensed Consolidated Statement of Changes in Stockholders' Equity; and (vi) the related Notes to the Condensed Consolidated Financial Statements. | ||||
| 104 # | Cover Page Interactive Data File, formatted in iXBRL and contained in Exhibit 101. | ||||
# Filed herewith.
† Certain portions of this exhibit have been omitted because they are not material and they are the type of information that the registrant treats as private or confidential.
70
EMERGENT BIOSOLUTIONS INC.
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| EMERGENT BIOSOLUTIONS INC. | ||
By: /s/ROBERT G. KRAMER Robert G. Kramer President, Chief Executive Officer and Director (Principal Executive Officer) | ||
| Date: November 5, 2021 | ||
By: /s/RICHARD S. LINDAHL Richard S. Lindahl Executive Vice President, Chief Financial Officer and Treasurer (Principal Financial and Accounting Officer) | ||
| Date: November 5, 2021 | ||
71

Certain identified information has been excluded from the exhibit because it is both (i) not material and (ii) is the type of information that the registrant treats as private or confidential. Double asterisks denote omissions. AWARD/CONTRACT 1. THIS CONTRACT IS A RATED ORDER UNDER OPAS (15 CFR 700) RATING PAGE OF PAGE 1 46 2. CONTRACT (Proc Inst Ident.) NO. HHSO100201600030C 3. EFFECTIVE DATE See Block 20C 4. ACQUISITION (PURCHASE REQUEST NO. OS165547 5. ISSUED BY CODE ASPR-BARDA 6. ADMINISTERED BY (If other ______ by) CODE ASPR BARDA ASPR-BARDA 200 Independence Ave., S.W. Room 640-G Washington DC 20201 ASPR-BARDA 200 Independence Ave., S.W. Room 638-G Washington DC 20201 7. NAME AND ADDRESS OF CONTRACTOR (No., Street, City Country, State and ZIP Code) EMERGENT PRODUCT DEVELOPMENT GAITHERSBURG INC. 1365869 EMERGENT PRODUCT DEVELOPMENT GAITHE 300 PROFESSIONAL DR # 100 GAITHERSBURG MD 208793419 8. DELIVERY FOB ORIGIN x OTHER (See below) 9. DISCOUNT(FOR PROMPT PAYMENT 10. SUBMIT INVOICES (4 copies unless otherwise specified) TO THE ADDRESS SHOWN IN ITEM CODE 1365869 FACILITY CODE 11. SHIP TO/MARK FOR CODE HHS/OS/ASPR 12. PAYMENT WILL BE MADE BY CODE PSC HHS/OS/ASPR 200 C St SW WASHINGTON DC 20201 Program Support Center 330 Independence Avenue SW Washington DC 20201 13. AUTHORITY FOR USING OTHER THAN FULL AND OPEN COMPETITION 10 USC 2304(c)( ) 41 USC 253(c) ( ) 14. ACCOUNTING AND APPROPRIATION DATA 2016.1990007.26201 15A. ITEM NO 15B. SUPPLIES/SERVICES 15C. QUANTITY 15D. UNIT 15E. UNIT PRICE 15F. AMOUNT Continued 15G. TOTAL AMOUNT OF CONTRACT $198,705,042.00 16. TABLE OF CONTENTS (X) SEC DESCRIPTION PAGE(S) (X) SEC. DESCRIPTION PART 1 – THE SCHEDULE PART II – CONTRACT CLAUSES X A SOLICITATION/CONTRACT FORM 1 X I CONTRACT CLAUSES 42 X B SUPPLIES OR SERVICES AND PRICES/COSTS 3 PART III – LIST OF DOCUMEMTS, EXHIBITS AND OTHER ATTACH X C DESCRIPTION/SPECS/WORK STATEMENT 10 X J LIST OF ATTACHMENTS 46 X D PACKAGING AND MARKING 11 PART IV – REPRESENTATIONS AND INSTRUCTIONS X E INSPECTION AND ACCEPTANCE 12 K REPRESENTATIONS, CERTIFICATIONS AND OTHER STATEMENTS OF OFFERORS X F DELIVERIES OR PERFORMANCE 13 X G CONTRACT ADMINISTRATION DATA 22 L INSTRS., CONDS., AND NOTICES TO OFFERORS X H SPECIAL CONTRACT REQUIREMENTS 26 M EVALUATION FACTORS FOR AWARD CONTRACTING OFFICER WILL COMPLETE ITEM 17 (SEALED BID OR NEGOTIATED PROCUREMENT) OR 18 (SEALED BID PROCUREMENT) AS APPLICABLE 17. X CONTRACTOR’S NEGOTIATED AGREEMENT (Contractor is required to sign this document and return 2 copies to issuing office.) Contractor agrees to furnish and deliver all items to perform all the services set forth or otherwise identified above and on any continuation sheets for the consideration stated herein. The rights and obligations of the parties to this contract shall be subject to and governed by the following documents (a) this award/contract, (b) the solicitation, if any, and (c) such provisions, representations, certifications, and specifications, as are attached or incorporated by reference herein. (Attachments are listed herein.) 18. SEALED BID AWARD (Contractor is not required to sign this document.) Your bid on Solicitation Number including the additions or changes made by you which additions or changes are set forth in full above, is hereby accepted as to the items listed above and on any continuation sheets. This award consummates the contract which consists of the following documents (a) the Government’s solicitation and your bid, and (b) this award/contract. No further contractual document is necessary. (Block 18 should be checked only when awarding a sealed-bid contract.) 19A. NAME AND TITLE OF SIGNER (Type or print) Adam Havey Adam Havey 20A. NAME OR CONTRACTING OFFICER BROOKE T. BERNOLD 19B. NAME OF CONTRACTOR Emergent BioSolutions BY /s/ Adam Havey (Signature of person authorized to sign) 19C. DATE SIGNED Sep 28, 2016 20B. UNITED STATES OF AMERICA BY /s/ Brooke Bernold (Signature of the Contracting Officer) 20C. DATE SIGNED 9/30/2016

AUTHORIZED FOR LOCAL REPRODUCTION Previous edition is NOT usable. STANDARD FORM 26 (Rev. 5/2011) Prescribed by GSA FAR (48 CFR) 53.214(a)

CONTINUATION SHEET REFERENCE NO. OF DOCUMENT BEING CONTINUE HHSO100201600030C PAGE OF 2 46 NAME OF OFFEROR OR CONTRACTOR EMERGENT PRODUCT DEVELOPMENT GAITHERSBURG INC. 1365869 ITEM NO. (A) SUPPLIES/SERVICES (B) QUANTITY (C) UNIT (D) UNIT PRICE (E) AMOUNT (F) Tax ID Number: [**] DUNS Number: [**] Next Generation Anthrax Vaccine Delivery: 09/27/2016 Appr. Yr.: 2016 CAN: 1990007 Object Class: 26201 FOB: Destination Period of Performance: 09/30/2016 to 09/29/2021 1 ASPR-16-06550 – CLIN 0001 and CLIN 0002 – Base contract to Emergent for the Manufacturing Development and Procurement of AV7909 (FDA Licensure and Approval/Initial Purchase, Storage, and Delivery of Product Obligated Amount: $198,705,042.00 198,705,042.00 2 CLIN 0001A – Phase II [**] Study of Studies Required by the FDA [**] Amount: $[**] (Option Line Item) 0.00 3 CLIN 0003 Post IV Marketing Commitments Amount: $[**] (Option Line Item) 0.00 4 CLIN 0004 (A-H) for Additional Surge Capacity (Based on [**] doses at the licensure price of $[**]) Amount: $[**] (Option Line Item) 0.00 AUTHORIZED FOR LOCAL REPRODUCTION OPTIONAL FORM 336 (4086) Sponsored by GSA FAR (48 CFR) 53 110

3 PART I – THE SCHEDULE SECTION B – SUPPLIES OR SERVICES AND PRICES/COSTS ARTICLE B.1. BRIEF DESCRIPTION OF SUPPLIES OR SERVICES Emergent Product Development Gaithersburg Inc. is developing NuThrax vaccine as a next generation anthrax vaccine. This vaccine is intended to mitigate anthrax as a bio threat against civilian populations suitable for Post-Exposure Prophylaxis (PEP) as a priority medical countermeasure. Under the base period-of-performance, Emergent will complete remaining development activities necessary to achieve licensure of the vaccine and manufacture and deliver vaccine product into the Strategic National Stockpile (SNS). The contract options may be exercised to perform additional studies necessary for licensure, support post-licensure commitments as required by the FDA, and procure additional treatment courses for the SNS. The Research and Development (R&D) effort will progress in specific stages that cover the base performance segment and several options, if necessary, as specified in this contract. The period of performance for the base period is 60 months. ARTICLE B.2. BASE PERIOD CLIN Period of Performance Supplies/ Services Total Est. Cost Fixed Fee (7%) Total Cost Plus Fixed Fee COST REIMBURSEMENT 0001 (Base) 09/30/2016 – 09/29/2021 Licensure, approval, and clearance of product through the FDA [**] [**] [**] FIRM FIXED PRICE CLIN Period of Performance Supplies/ Services Units (# of Doses) Unit Price ($) Total ($) 0002 (Base) 09/30/2016 – 09/29/2021 Initial Purchase, Storage, and Delivery of Product 2,000,000 [**] [**] Total CLINS 1&2 09/30/2016 – 09/29/2021 See Above Descriptions $198,705,042 (Funded) ARTICLE B.3. OPTION PRICES CLIN Period of Performance Supplies/ Services Total Est. Cost Fixed Fee Total Cost Plus Fixed Fee ($) COST REIMBURSEMENT

4 0001A (Option Quantity) [**] Phase II [**] Study or studies required by the FDA [**] [**] [**] [**] CLIN Period of Performance Supplies/ Services Total Est. Cost Fixed Fee Total Cost Plus Fixed Fee ($) FIXED PRICE 0003 (Option Quantity) [**] Phase IV post marketing commitments /Requirements (This is an option that may or may not be exercised during the base period as determined by the need and as established by the FDA) N/A N/A [**] CLIN Period of Performance Supplies/ Services Units (# of Product) FY 2018 Unit Price ($) Total ($) 0004A (Option Quantity) [**] Additional Surge Capacity (EUA) 7,500,000 to [**] [**] [**] 0004B (Option Quantity) [**] Additional Surge Capacity (Licensure) 7,500,000 to [**] [**] [**] 0004C (Option Quantity) [**] Additional Surge Capacity (EUA) [**] [**] [**] 0004D (Option Quantity) [**] Additional Surge Capacity (Licensure) [**] [**] [**] 0004E (Option Quantity) [**] Additional Surge Capacity (EUA) [**] [**] [**] 0004F (Option Quantity) [**] Additional Surge Capacity (Licensure) [**] [**] [**] 0004G (Option Quantity) [**] Additional Surge Capacity (EUA) [**] [**] [**] 0004H (Option Quantity) [**] Additional Surge Capacity (Licensure) [**] [**] [**] ARTICLE B.4. LIMITATIONS APPLICABLE TO DIRECT COSTS a. Items Unallowable Unless Otherwise Provided Notwithstanding the clause FAR 52.216-7, Allowable Cost and Payment, incorporated in this contract, the costs of the following items or activities shall be unallowable as direct costs unless

5 authorized in writing in advance by the Contracting Officer: 1. Acquisition, by purchase or lease, of any interest in real property; 2. Special rearrangement or alteration of facilities; 3. Purchase or lease of any item of general purpose office furniture or office equipment regardless of dollar value. (General purpose equipment is defined as any items of

6 personal property which are usable for purposes other than research, such as office equipment and furnishings, pocket calculators, etc.); 4. Travel to attend general scientific meetings; 5. Unapproved foreign travel; 6. Consultant costs; 7. Subcontracts; 8. Patient care costs; 9. Accountable Government property (defined as both real and personal property with an acquisition cost of $1,000 or more and a life expectancy of more than two years) and “sensitive items” (defined as items of personal property, supplies and equipment that are highly desirable and easily converted to personal use), regardless of acquisition value. 10. Printing Costs (as defined in the Government Printing and Binding Regulations). 11. Light Refreshment and Meal Expenditures - Requests to use contract funds to provide light refreshments and/or meals to either federal or nonfederal employees must be submitted to the Contracting Officer’s Representative (COR), with a copy to the Contracting Officer, at least six (6) weeks in advance of the event and are subject to “HHS Policy on Promoting Efficient Spending: Use of Appropriate Funding for Conferences and Meeting, Food and Promotional Items and Printing and Publications.” The request shall contain the following information: (a) name, date, and location of the event at which the light refreshments and/or meals will be provided; (b) a brief description of the purpose of the event; (c) a cost breakdown of the estimated light refreshments and/or meals costs; (d) the number of nonfederal and federal attendees receiving light refreshments and/or meals; and (e) if the event will be held at a government facility. 12. Meeting room or conference space used for face to face meetings with USG staff in the performance of this contract. Justification for why the meeting cannot be held at a government facility must be provided. COA requests must be made at least (2) two weeks prior to meeting date. 13. Clinical Trial Insurance b. Travel Costs 1. During the Base Period total expenditures for travel (transportation, lodging, subsistence, and incidental expenses) incurred by the Prime Contractor in direct performance of this contract shall not exceed $[**] without prior advance written approval by the Contracting Officer. Costs must be consistent with FAR 52.247-63 – Preference for U.S.- Flag Air Carriers. 2. The Contactor shall invoice and be reimbursed for all travel costs in accordance with FAR 31.205-46, Contracts with Commercial Organizations, Travel Costs. 3. Requests for foreign travel must be submitted at least six weeks in advance and shall contain the following: (i) Meeting(s) and place(s) to be visited, with costs and dates; (ii) Names(s) and title(s) of Contractor personnel to travel and their functions in the contract project; (iii) Contract purpose to be served by the travel; (iv) How travel of Contractor personnel will benefit and contribute to accomplishing the contract project, or will otherwise justify the expenditure of AMCG contract funds;

7 (v) How such advantages justify the costs for travel and absence from the project of more than one person if such are suggested; and (vi) What additional functions may be performed by the travelers to accomplish other purpose of the contact and thus further benefit the project. ARTICLE B.5. ADVANCE UNDERSTANDINGS a. Subcontracts and Consultants Award of any FFP subcontract or FFP consulting agreement in excess of $150,000 or any cost reimbursement subcontract or consulting agreement shall not proceed without the prior written consent of the Contracting Officer via a Contracting Officer Authorization (COA) Letter. COA letters will only be issued upon review of the supporting documentation required by FAR Clause 52.244-2, Subcontracts. After receiving written consent of the subcontract by the Contracting Officer, a copy of the signed, executed subcontract and consulting agreement shall be provided to the Contracting Officer within ten (10) calendar days of full execution. b. Site Visits, Inspections and General Audits At the discretion of the USG and independent of activities conducted by the Contractor, with 48 hours’ notice to the Contractor, the USG reserves the right to conduct site visits and inspections on an as needed basis, including collection of product samples and intermediates held by the Contractor, or subcontractor. In case of subcontractor visits and inspections that are independent of activities conducted by the Contractor, the USG shall demonstrate cause for such visit and/or inspection. All costs reasonably incurred by the Contractor and subcontractor for such visit and/or inspection shall be allowable costs. The Contractor shall coordinate these visits and shall have the opportunity to accompany the USG on any such visits. Under time-sensitive or critical situations, the USG reserves the right to suspend the 48 hour notice to the Contractor. If the Government, Contractor, or other party identifies any issues during an audit, the Contractor shall capture the issues, identify potential solutions, and provide a report to the Government for review and acceptance. • If issues are identified during the audit, Contractor shall submit an issue report to the CO and COR within 10 business days detailing the finding and corrective action(s) of the audit. • COR and CO will review the issues report and provide a response to the Contractor within 10 business days. • Once corrective action is completed, the Contractor will provide a final report to the CO and COR within a time frame negotiated with the COR in writing after review of the issues report. c. QA Audits BARDA reserves the right to participate in QA audits. Upon completion of the QA audit the Contractor shall provide a report capturing the findings, results, and next steps in proceeding with any potential subcontractors. If action is requested for a subcontractor, detailed corrective and preventative plans for addressing areas of non-conformance to ICH and FDA regulations for GLP, GMP, or GCP guidelines, as identified in the audit report, must be provided to BARDA for review and acceptance. The Contractor shall provide responses from the subcontractors to address these concerns and plans for corrective action execution. • Contractor shall notify CO and COR of upcoming, ongoing, or recent audits/site visits of subcontractors as part of weekly communications. • Contractor shall notify the COR and CO within 5 business days of report completion. The Contractor shall complete the report within 60 days of the audit/site visit, or as negotiated with the COR in writing dependent upon the audit findings.

8 d. Man-in-Plant At the discretion of the Government and seven calendar (7) days advance notice to the Contractor in writing from the Contracting Officer, the Government may place a man-in- plant in the Contractor’s facility, who shall be subject to the Contractor’s policies and procedures regarding security and facility access at all times while in the Contractor’s facility. As determined by federal law, no Government representative shall publish, divulge, disclose, or make known in any manner, or to any extent not authorized by law, any information coming to him in the course of employment or official duties, while stationed in a contractor plant. e. Emergency Use Authorization (EUA) The Contractor shall be responsible for generating the data to support the USG’s filing of a Pre-Emergency Use Authorization (Pre-EUA) package for use of the product prior to FDA licensure or approval during a declared emergency, declared potential emergency, or identification of material threat under an Emergency Use Authorization (EUA). The Contractor commits to supporting the potential use of the product under a pre-EUA package as submitted by BARDA or the CDC/SNS. The Contractor shall supply BARDA or the CDC/SNS with the data needed to support such a submission, including expanded access INDs, right to hold product, right of reference to the Contractor's Investigational New Drug (IND), or other application that contains the supporting data. The Contractor shall address any FDA comments on all pre-EUA packages as applicable. The Contractor shall maintain and update, as required by the FDA, all required regulatory documentation (investigator brochure, regulatory binder, etc.), that will be used to support use under EUA and approval/licensure. Any product which has not received FDA approval or licensure, but has completed submission of a Pre-EUA package deemed acceptable by the FDA and has met the two (2) criteria listed below may be considered for procurement at the discretion of the USG. The Contractor would be required to demonstrate the two (2) essential criteria listed below for consideration of procurement of any unapproved products by seeking a COA. The COA shall include a product delivery schedule for consideration and documentation of the following: • Substantial evidence, including a validated process, of the Contractor’s ability to manufacture a product that would be identical to the commercial scale as required for product approval or licensure. A clear understanding of the outstanding risks, if any, for approval or licensure must be demonstrated. • Completion of non-clinical and clinical studies with substantial evidence of safety and efficacy for the indicated use. A list of outstanding activities and targets for completion, adverse events/safety profile which do not pose unusual risks or challenges for FDA approval or licensure shall be provided. A tentative delivery schedule of product delivery to the inventory (acceptable as in the Quality Agreement) shall be required as part of the COA. The delivery schedule shall be updated periodically as necessary. For information concerning EUA, please consult http://www.fda.gov/RegulatoryInformation/Guidances/ucm125127 and http://www.fda.gov/EmergencyPreparedness/Counterterrorism/MedicalCountermeasures/ MCMLegalRegulatoryandPolicyFramework/ucm182568.htm

9 f. Sharing of contract deliverables within United States Government (USG) In an effort to build a robust medical countermeasure pipeline through increased collaboration, BARDA may share technical deliverables with USG entities responsible for Medical Countermeasure Development. In accordance with recommendations from the Public Health Emergency Medical Countermeasure Enterprise Review, agreements established in the Integrated Portfolio’s Portfolio Advisory Committee (PAC) Charter, and agreements between BARDA and the Department of Defense and the National Institutes of Health, BARDA may share technical deliverables and data created in the performance of this contract with colleagues within the Integrated Portfolio. This advance understanding does not authorize BARDA to share financial information outside HHS. The Contractor is advised to review the terms of FAR 52.227-14, Rights in Data – General, regarding the Government’s rights to deliverables submitted during performance as well as the Government’s rights to data contained within those deliverables. g. Overtime Compensation No overtime (premium) compensation is authorized under the subject contract. Billing of actual hours should be limited to total productive hours in a month. h. Option CLINS If procurement for CLIN 4 occurs after FY 2018, the following chart illustrates the dose prices to be used: Units (# of Doses) FY 2019 Unit Price ($) FY 2020 Unit Price ($) FY 2021 Unit Price ($) 7,500,000 to [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] The USG reserves the right to re-negotiate the option CLINS based on availability of funds and feedback received from the FDA. i. Contract Number Designation On all correspondence submitted under this contract, the Contractor agrees to clearly identify the contract number that appears on the face page of the contract as follows: HHSO100201600030C

10 j. Quality Agreement The Quality Agreement shall define, establish, and document the responsibilities of both the Contractor and the USG (i.e. – CDC/SNS-Quality Control and BARDA) for event- driven and product shipping, receiving, acceptance into the inventory and/or custody by the USG. These documents shall be drafted, approved, and signed by all parties prior to the commencement of product procurement and acceptance, transport and custody of the product under the CDC/SNS. The Contractor shall provide documentation and resolution for all concerns raised by USG and commits to cooperation in execution of this agreement.

11 SECTION C - DESCRIPTION/SPECIFICATIONS/WORK STATEMENT ARTICLE C.1. STATEMENT OF WORK Independently and not as an agent of the Government, the Contractor shall furnish all the necessary services, qualified personnel, material, equipment, and facilities not otherwise provided by the Government as needed to perform the Statement of Work dated September 30, 2016 set forth in SECTION J - List of Attachments, attached hereto and made a part of the contract. ARTICLE C.2. REPORTING REQUIREMENTS See Section F for specific reporting requirements. All reports required herein shall be submitted in electronic format. All paper/hardcopy documents/reports submitted under this contract shall be printed or copied, double-sided, on at least 30 percent post-consumer fiber paper, whenever practicable, in accordance with FAR 4.302(b). ARTICLE C.3. TWICE MONTHLY CONFERENCE CALLS A conference call between the Contracting Officer’s Representative (COR) and the Contractor’s Project Leaders/delegates and designees shall occur twice-monthly or as directed by the Contracting Officer and Contracting Officer’s Representative. During this call the Contractor’s Project Leaders/delegates and designees will discuss the activities since the last call, any problems that have arisen and the activities planned until the next call takes place. The Contractor’s Project Leaders/delegates may choose to include other key personnel on the conference call to give detailed updates on specific projects or this may be requested by the Contracting Officer’s Representative. ARTICLE C.4. PROJECT MEETINGS The Contractor shall participate in Project Meetings to coordinate the performance of the contract, as requested by the Contracting Officer’s Representative. These meetings may include face-to- face meetings with AMCG/BARDA in Washington, D.C. and at work sites of the Contractor. Such meetings may include, but are not limited to, meetings of the Contractor to discuss study designs, site visits to the Contractor’s facilities, and meetings with the Contractor and HHS officials to discuss the technical, regulatory, and ethical aspects of the program. Subject to the data rights provisions in this contract, the Contractor will provide data, reports, and presentations to groups of outside experts and USG personnel as required by the Contracting Officer and Contracting Officer’s Representative in order to facilitate review of contract activities.

12 SECTION D – PACKAGING, MARKING AND SHIPPING All deliverables required under this contract shall be packaged, marked and shipped in accordance with Government specifications. At a minimum, all deliverables shall be marked with the date, contract number and Contractor name. The Contractor shall guarantee that all required materials shall be delivered in immediate usable and acceptable condition.

13 SECTION E – INSPECTION AND ACCEPTANCE The Contracting Officer or the duly authorized representative will perform inspection and acceptance of materials and services to be provided under this contract. For the purpose of this SECTION E, the designated Contracting Officer’s Representative (COR) is the authorized representative of the Contracting Officer. The COR will assist in resolving technical issues that arise during performance. The COR however is not authorized to change any contract terms or authorize any changes in the Statement of Work or modify or extend the period of performance, or authorize reimbursement of any costs incurred during performance. The Contractor is advised to review FAR 52.243-1 Changes – Fixed Price Contracts Alternate V and FAR 52.243-2 Changes-Cost reimbursement contracts Alternative V, which is incorporated by reference into this contract in ARTICLE I.1. Inspection and acceptance will be performed at: Office of Acquisition Management, Contracts, and Grants (AMCG) Office of the Assistant Secretary for Preparedness and Response U.S. Department of Health and Human Services 200 C St. SW Washington, D.C. 20024 Acceptance may be presumed unless otherwise indicated in writing by the Contracting Officer or the duly authorized representative within 30 days of receipt. The contract incorporates the following clause by reference with the same force and effect as if it were given in full text. Upon request, the Contracting Officer will make its full text available. FAR 52.246-4, Inspection of Services - Fixed Price (August 1996) FAR 52.246-5, Inspection of Services - Cost-Reimbursement (April 1984) FAR 52.246-9, Inspection of Research and Development (Short Form) (April 1984) FAR 52.246-16, Responsibility for Supplies (April 1984)

14 SECTION F – DELIVERIES OR PERFORMANCE ARTICLE F.1. PERIOD OF PERFORMANCE The period of performance for this contract shall be from September 30, 2016 through September 29, 2021. The period of performance for the base period of this contract shall be consistent with the dates set forth in SECTION B. If the Government exercises option(s), the period of performance will be extended as described under in SECTION B of this contract. ARTICLE F.2. REPORTING REQUIREMENTS In all cases the reports are intended to provide sufficient detail to understand the Contractor's approach and progress to addressing the technical requirements. The reports supplement, and do NOT replace, routine (i.e. daily) communication between the COR and project manager and/or their designee(s) regarding project plans and progress. A. Monthly Progress Report This report shall include a description of the activities during the reporting period and the activities planned for the ensuing reporting period. The first reporting period consists of the first full month of performance plus any fractional part of the initial month. Thereafter, the reporting period shall consist of each calendar month. The Contractor shall submit a Monthly Progress Report on or before the 15th calendar day following the last day of each reporting period and shall include the following: Title Page: The title page for this report shall include the contract number and title; the type of report and period that it covers; the Contractor's name, address, telephone number, fax number, and e-mail address; and the date of submission. Distribution List: A list of individuals receiving the Technical Progress report. Progress: SECTION I - An introduction covering the purpose and scope of the contract effort. SECTION II Part A: SUMMARY - A description or table summarizing ongoing activities. SECTION II Part B: MANAGEMENT AND ADMINISTRATIVE UPDATE – This section shall include a description of all meetings, conference calls, etc. that have taken place during the reporting period. Include progress on administration and management issues (e.g. evaluating and managing subcontractor performance and personnel changes). Please include all Quality Management System, Quality Control, and Quality Assurance updates as part of this report or as requested by the COR. SECTION II Part C: TECHNICAL PROGRESS – This section shall document the results of work completed and costs incurred during the period covered in relation to the proposed progress, effort, and budget. The report shall be in sufficient detail to explain comprehensively the results achieved. SECTION II Part D: ISSUES – This section shall include a description of problems encountered and proposed corrective action; differences between planned and actual progress; why the differences have occurred and what corrective actions are planned; and if a project activity is delinquent, then what corrective action steps are planned. Revised timelines shall be provided. SECTION II Part E: PROPOSED WORK – This section shall include a summary of work proposed as a rolling three (3) month forecast for the next reporting period, by a certain date, and by whom.

14 SECTION II Part F: MANUFACTURING AND SUPPLY CHAIN MANAGEMENT – This section shall include a summary of the manufacturing and supply-chain related activities. Also include in this section updates to the production plan, capacity projections, stability results, inventory and shipment/distribution information. Invoices: Summary of any invoices submitted during the reporting period. A Monthly Progress Report will not be required in the same months that Annual or Final Technical Progress Reports are due. B. Annual Progress Report This report shall include a summation of the activities during the reporting period, and the activities planned for the ensuing reporting period. The first reporting period consists of the first full year of performance plus any fractional part of the initial year. Thereafter, the reporting period shall consist of each calendar year. The Contractor shall submit an Annual Progress Report on or before the 30th calendar day following the last day of each reporting period and shall include the following: Title Page: The title page for this report shall include the contract number and title; the type of report and period that it covers; the Contractor's name, address, telephone number, fax number, and e-mail address; and the date of submission. Distribution List: A list of individuals receiving the Technical Progress report. Progress: SECTION I - An introduction covering the purpose and scope of the contract effort. SECTION II Part A: SUMMARY - A description or table summarizing ongoing activities. SECTION II Part B: MANAGEMENT AND ADMINISTRATIVE UPDATE – This section shall include a description of all meetings, conference calls, etc. that have taken place during the reporting period. Include progress on administration and management issues (e.g. evaluating and managing subcontractor performance and personnel changes). Please include all Quality Management System, Quality Control, and Quality Assurance updates as part of this report or as requested by the COR. SECTION II Part C: TECHNICAL PROGRESS – This section shall document the results of work completed and costs incurred during the period covered in relation to proposed progress, effort, and budget. The report shall be in sufficient detail to explain comprehensively the results achieved. SECTION II Part D: ISSUES – This section shall include a description of problems encountered and proposed corrective action; differences between planned and actual progress; why the differences have occurred and what corrective actions are planned; and if a project activity is delinquent, then what corrective action steps are planned. Revised timelines shall be provided. SECTION II Part E: PROPOSED WORK – This section shall include a summary of work proposed as an annual forecast for the next reporting period, by a certain date, and by whom. SECTION II Part F: MANUFACTURING AND SUPPLY CHAIN MANAGEMENT – This section shall include a summary of the manufacturing and supply-chain related activities. Also include in this section updates to the production plan, capacity projections, stability results, inventory and shipment/distribution information. Invoices: Summary of any invoices submitted during the reporting period.

15 An Annual Progress Report will not be required for the period when the Final Technical Progress Report is due. C. Draft Final Report and Final Report These reports are to include a summation of the work performed and results obtained for execution of various studies or technical work packages during the entire contract period of performance. This report shall be in sufficient detail to describe comprehensively the results achieved. The Draft Final Progress Report shall be due forty-five (45) calendar days prior to the expiration date of the contract and the Final Progress Report is due on or before the expiration date of the contract. The report shall conform to the following format: Title Page: The title for these reports shall include the contract number and title; the type of report and period that it covers; the Contractor's name, address, telephone number, fax number, and e-mail address; and the date of submission. Distribution List: A list of individuals receiving the Technical Progress report. Progress: SECTION I: EXECUTIVE SUMMARY - Summarize the purpose and scope of the contract effort including a summary of the major accomplishments relative to the specific activities set forth in the Statement of Work. SECTION II: RESULTS - A detailed description of the work performed and the results obtained including all expenses for the entire contract period of performance. D. FDA Regulatory Agency Correspondence, Meeting Summaries, and Submissions. a. Within five business days of any formal meeting with the FDA or other regulatory agency, the Contractor shall forward the initial draft minutes to BARDA. The Contractor shall forward the final minutes when available. b. Within five business days of any informal meeting with the FDA or other regulatory agency, the Contractor shall provide a formal contact report to BARDA. The Contractor shall forward the final minutes when available and if applicable. c. The Contractor shall forward the dates and times of any formal meeting with the FDA and other regulatory agencies to BARDA as soon as the meeting times are known and make arrangements for appropriate BARDA staff to attend the meetings. d. The Contractor shall provide BARDA the opportunity to review and comment upon any documents to be submitted to the FDA or other regulatory agency. The Contractor shall provide BARDA with five (5) business days in which to review and provide comments back to the Contractor prior to the Contractor’s submission to the FDA. e. The Contractor shall make Standard Operating Procedures (SOPs) available upon request from COR. f. The Contractor shall provide raw data and/or specific analysis of data generated with USG funds upon request from the COR. g. The Contractor shall notify the Contracting Officer’s Representative and Contracting Officer within 24 hours of all site visits/audits conducted by the FDA or any other regulatory agency. The Contractor shall provide the USG with an exact copy (non-redacted) of the FDA Form 483 and the Establishment Inspection Report (EIR). The Contractor shall provide the Contracting Officer’s

16 Representative and Contracting Officer copies of the plan for addressing areas of non-conformance to FDA regulations for GLP guidelines as identified in the audit report, status updates during the plans execution, and a copy of all final responses to the FDA. The Contractor shall also provide redacted copies of any FDA audits received from subcontractors that occur as a result of this contract or for this product. The Contractor shall make arrangements with the COR for the appropriate BARDA representative(s) to be present during the final debrief by the regulatory inspector. E. Other Requirements/Deliverables a. Integrated Master Project Plan The Contractor shall provide an Integrated Master Project Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to annual deliverables and Work Breakdown Structure (WBS) elements. Attention shall be placed on providing sufficient turnaround time for the USG (BARDA, FDA, and CDC) for review of critical documentation. The Contractor shall integrate to demonstrate interdependencies among all CLINS. The Integrated Master Project Plan shall be incorporated into any potential contract and will be used to monitor performance of the contract. This report shall be due within 90 days of contract award. Updates shall be due as requested by the COR or Alternate COR. i. Critical Path Milestones The Integrated Master Project Plan shall outline key, critical path milestones, with “Go/No Go” decision criteria (entrance and exit criteria for each phase of the project). This report shall be due within 90 days of contract award. Updates shall be due as requested by the COR or Alternate COR. ii. Work Breakdown Structure The USG has provided a Contract Work Breakdown Structure (CWBS) template (See http://www.phe.gov/about/amcg/contracts/Pages/toolkit.aspx) and the Contractor shall further delineate the CWBS to Level 5 as part of their Integrated Master Project Plan. The WBS shall be discernable and consistent. BARDA may require Contractor to furnish WBS data at the work package level or at a lower level if there is significant complexity and risk associated with the task. This report shall be due within 90 days of contract award. Updates shall be due as requested by the COR or Alternate COR. iii. Risk Mitigation Plan/Matrix The Contractor shall develop and maintain a risk management plan that highlights potential problems and/or issues that may arise during the life of the contract, their impact on cost, schedule and performance, and appropriate remediation plans. This plan shall reference relevant WBS/SOW elements where appropriate. The USG has provided a Risk Mitigation Matrix template (See http://www.phe.gov/about/amcg/contracts/Pages/toolkit.aspx) to be completed by any prospective Contractor. This report shall be due within 90 days of contract award. Updates shall be due as requested by the COR or Alternate COR. b. Technology Packages Technology packages developed under the contract that includes complete protocols must be submitted at the request of the BARDA Contracting Officer’s Representative. See FAR clauses 52.227- 11, Patent Rights-Ownership by the Contractor, and 52.227-14, Rights in Data. This report shall be due upon request from the COR or Alternate COR.

17 c. Experimental Protocols The Contractor shall submit to the COR all protocols and associated study/experiment/test plans prior to the execution of any non-clinical animal study or clinical study for BARDA approval or upon request by the COR or Alternate COR when required. Approval must be provided in writing by the COR or Alternate COR prior to the execution of the study. d. Annual/Final Invention Report All reports and documentation required by FAR Clause 52.227-11, Patent Rights- Ownership by the Contractor, including, but not limited to, the invention disclosure report, the confirmatory license, and the Government support certification. An Annual Invention Report shall be due on or before the 30th calendar day after the completion of each reporting period. A Final Invention Report (see FAR 27.303 (b)(2)(ii)) shall be due on or before the expiration date of the contract. If no invention is disclosed or no activity has occurred on a previously disclosed invention during the applicable reporting period, a negative report shall be submitted to the Contracting Officer. e. Publications Any manuscript or scientific meeting abstract containing data generated under this contract must be submitted to COR for review prior to submission. Publications are due within 10 business days for manuscripts and 5 business days for abstracts prior to public release. f. Press Releases The Contractor agrees to accurately and factually represent the work conducted under this contract in all press releases. The Contractor shall ensure the Contracting Officer has received and approved an advanced copy of any press release not less than five (5) business days prior to the issuance of any potential press release. g. Incident Security Report The Contractor shall report to the government any activity; or incident that is in violation of established security standards; or indicates the loss or theft of government products. Reports shall be due within 24 hours after occurrence of an activity or incident. h. Security Plan The Contractor shall submit a draft security plan within 90 days of contract award. A detailed security plan with any updates shall be submitted for approval at least three (3) months prior to the initiation of product procurement with proper documentation. The Contractor shall cooperate with USG representatives to develop a sustainable security plan to ensure continued security of the premises. Security plan updates are required when an incident security report has been filed. F. Earned Value Management System Plan a. Earned Value Management System Plan: Subject to the requirements under HHSAR Clause 352.234-3, the Contractor shall use principles of Earned Value Management System (EVMS) in the management of this contract (include this plan as part of the monthly, annual, and final reports). The Seven Principles are: I. Plan all work scope for the program to completion. II. Break down the program work scope into finite pieces that can be assigned to a responsible person or organization for control of technical, schedule, and cost objectives.

18 III. Integrate program work scope, schedule, and cost objectives into a performance measurement baseline plan against which accomplishments may be measured. Control changes to the baseline. IV. Use actual cost incurred and recorded in accomplishing the work performed. V. Objectively assess accomplishments at the work performance level. VI. Analyze significant variances from the plan, forecast impacts, and prepare an estimate at completion based on performance to date and work to be performed. VII. Use earned value information in the company’s management processes. VIII. Elements of EVMS shall be applied to all CLINs as part of the Integrated Master Project Plan, the Contractor shall submit a written summary of the management procedures that it will establish, maintain and use to comply with EVMS requirements. b. Performance Measurement Baseline Review (PMBR): The Contractor shall submit a PMBR plan electronically via email to the CO and COR for a PMBR to occur within 90 days of contract award. At the PMBR, the Contractor and BARDA shall mutually agree upon the budget, schedule and technical plan baselines (Performance Measurement Baseline). These baselines shall be the basis for monitoring and reporting progress throughout the life of the contract. The PMBR is conducted to achieve confidence that the baselines accurately capture the entire technical scope of work, are consistent with contract schedule requirements, are reasonably and logically planned, and have adequate resources assigned. The goals of the PMBR are as FOLLOWS: I. Jointly assess areas such as the Contractor’s planning for complete coverage of the SOW, logical scheduling of the work activities, adequate resources, and identification of inherent risks. II. Confirm the integrity of the Performance Measurement Baseline (PMB). III. Foster the use of EVM as a means of communication. IV. Provide confidence in the validity of Contractor reporting V. Identify risks associated with the PMB. VI. Present any revised PMBs for approval. VII. Present an Integrated Master Schedule: The Contractor shall deliver an initial program level Integrated Master Schedule (IMS) that rolls up all time-phased WBS elements down to the activity level. This IMS shall include the dependencies that exist between tasks. This IMS will be agreed to and finalized at the PMBR. DI- MGMT-81650 may be referenced as guidance in creation of the IMS (see http://www.acq.osd.mil/pm/). VIII. Present the Risk Management Plan.

19 c. Integrated Master Schedule The Contractor shall provide a program Integrated Master Schedule (IMS) with monthly status updates (e.g. % complete with program tasks). Initial IMS due thirty (30) days after award. Monthly status updates are due the 20th day of the month after the end of each month. The Integrated Master Schedule shall be incorporated into the contract, and shall be used to monitor performance of the contract. The Contractor shall include the key milestones and Go/No Go decision gates. The Contractor shall include BARDA Portfolio Management Milestones (See the AMCG Business Toolkit for a description and sample (http://www.phe.gov/about/amcg/contracts/Pages/toolkit.aspx) in their IMS and provide monthly updates within their IMS. This IMS shall include the following fields at a minimum; baseline start and finish, forecast start and finish, actual start and finish, predecessor and/or successor. The Contractor shall deliver the Integrated Master Schedule, viewed at the work package level in MS Project file format d. Earned Value Contract Performance Report (EV-CPR) a. The Offeror shall deliver an Earned Value Contract Performance Report (CPR) on a monthly basis per the instruction in DI-MGMT-81466A (see http://www.acq.osd.mil/pm/). The Contractor shall provide Format 1, Format 3, and Format 5 only. Format 1 will be reported at the Work Breakdown Structure level 3 agreed to by BARDA and the Contractor. b. EV Variance thresholds will be negotiated with the Contractor post-award but for planning purposes will likely be (+/- 10% and $30,000). In conjunction with the CPR, the Contractor shall provide a monthly update to the IMS with up to date performance data and shall include actual start/finish and projected start / finish dates. c. The supplemental monthly CAP report shall contain, at the work package level, time phased budget (budgeted cost of work scheduled (BCWS)), earned value (budgeted cost of work performed (BCWP)), and actual costs of work performed (ACWP) as captured in the Contractor’s EVM systems. d. The Contractor and BARDA shall participate in regular meetings to coordinate and oversee the contracting effort as requested by the COR. Such meetings may include, but are not limited to, site visits to the Contractor’s and/or subcontractor’s facilities, meetings with individual Contractors and other HHS officials to discuss the technical, regulatory, and ethical aspects of the program. The Contractor shall provide data, reports, and presentations to groups of outside experts and USG personnel and Government-contracted subject matter experts as required by the BARDA COR in order to facilitate review of contract activities. e. The Contractor shall provide a list of individuals to serve as primary and secondary points of contact who will be available 24 hours a day, seven days a week, to be notified in case of a public health emergency. ARTICLE F.3. DELIVERIES Successful performance of the final contract shall be deemed to occur upon performance of the work set forth in the Statement of Work dated September 30, 2016, set forth in SECTION J - List of Attachments of this contract and upon delivery and acceptance by the Contracting Officer, or the duly authorized representative, of the following items in accordance with the stated delivery schedule below:

20 Item No. Description Addresses Deliverable Schedule 1 Monthly Progress Report CO: (1) electronic copy COR: (1) electronic copy Reports are due on or before the 15th of each month following the end of each reporting period. 2 Annual Progress Report CO: (1) electronic copy COR: (1) electronic copy Reports are due on or before the 30th calendar day following the end of each reporting period. 3 Draft Final Progress Report CO: (1) electronic copy COR: (1) electronic copy Report is due 45 Calendar days prior to the expiration date of the contract. 4 Final Progress Report CO: (1) electronic copy COR: (1) electronic copy Report is due on or before the expiration date of the contract. 5 FDA/ Regulatory Agency Correspondence and Meeting Summaries COR: (1) electronic copy Reports are due within 5 business days of each meeting for Contractor’s minutes, upon receipt of minutes from FDA/ regulatory agency, and upon request from the COR or Alternate COR. 6 Integrated Master Project Plan -Critical Path Milestones - Work Breakdown Structure - Risk Mitigation Plan/Matrix COR: (1) electronic copy Report is due within 90 days of contract award. Updates are due as requested by the COR or Alternate COR. 7 Technology Packages COR: (1) electronic copy Upon request from the COR or Alternate COR. 8 Experimental Protocols for non- clinical animal studies or clinical studies COR: (1) electronic copy Upon request from the COR or Alternate COR. Written approval from the COR or Alternate COR is required prior to the execution of the study. 9 Annual/Final Invention Report CO: (1) electronic copy COR: (1) electronic copy An Annual Invention Report is due on or before the 30th calendar day after the completion of each reporting period. A Final Invention Report is due on or before the expiration date of the

21 contract. 10 Publications COR: (1) electronic copy Reports are due within 10 business days for manuscripts and 5 business days for abstracts. 11 Press Releases CO: (1) electronic copy COR: (1) electronic copy Reports/Notices are due for approval to the CO not less than five (5) business days prior to the issuance of any potential press release. 12 Incident Security Report CO: (1) electronic copy COR: (1) electronic copy Reports are due within 24 hours after occurrence of an activity or incident. 13 Security Plan CO: (1) electronic copy COR: (1) electronic copy Draft report is due within 90 days of contract award. Updates are due at least 3 months prior to product procurement or as requested by the COR or Alternate COR. 14 Earned Value Management Requirements CO: (1) electronic copy COR: (1) electronic copy As detailed in Section F.2 Reporting Requirements, subpart -F. Email Addresses: CO – [**] COR – [**] ARTICLE F.4. FEDERAL ACQUISITION REGULATION CLAUSES INCORPORATED BY REFERENCE, FAR 52.252-2 (FEBRUARY 1998) This contract incorporates the following clause(s) by reference, with the same force and effect as if it were given in full text. Upon request, the Contracting Officer will make its full text available. The full text of each clause may be accessed electronically at this address: http://www.acquisition.gov/far. FAR 52.242-15, Stop Work Order (August 1989) FAR 52.242-15, Alternate 1 (April 1984) is applicable to this contract.

22 SECTION G - CONTRACT ADMINISTRATION DATA ARTICLE G.1. CONTRACTING OFFICER The following Contracting Officer (CO) will represent the Government for the purpose of this contract: [**] DHHS/OS/ASPR/AMCG 200 C St. Washington, D.C. 20024 a. The Contracting Officer (CO) is the only individual who can legally commit the Government to the expenditure of public funds. No person other than the CO can make any changes to the terms, conditions, general provisions, specifications or other requirements of this contract. b. The Contracting Officer (CO) is the only person with authority to act as agent of the Government under this contract. Only the CO has authority to: (1) direct or negotiate any changes in the statement of work; (2) modify or extend the period of performance; (3) change the delivery schedule; (4) authorize reimbursement to the Contractor for any costs incurred during the performance of this contract; or (5) otherwise change any terms and conditions of this contract. c. No information, other than that which may be contained in an authorized modification to this contract duly issued by the CO, shall be considered grounds for deviation from this contract. d. The Government may unilaterally change its CO designation. ARTICLE G.2. CONTRACTING OFFICER'S REPRESENTATIVE (COR) The following Contracting Officer's Representative (COR) will represent the Government for the purpose of this contract: [**] Contracting Officer's Representative Biomedical Advanced Research and Development Authority (BARDA) Office of the Assistant Secretary for Preparedness and Response Department of Health and Human Services [**] Mailing Address: 200 C St. Washington, D.C. 20024 Alternate COR: [**] Alternate Contracting Officer’s Representative (COR) Biomedical Advanced Research and Development Authority (BARDA) Office of the Assistant Secretary for Preparedness and Response Department of Health and Human Services [**] Mailing Address: 200 C St. Washington, D.C. 20024

23 The COR is responsible for: a. Monitoring the Contractor's technical progress, including the surveillance and assessment of performance and recommending to the Contracting Officer changes in requirements; b. Assisting the Contracting Officer in interpreting the statement of work and any other technical performance requirements; c. Performing technical evaluation as required; d. Performing technical inspections and assisting the Contracting Officer in acceptances of deliverables required by this contract; and e. Assisting in the resolution of technical problems encountered during performance. f. The Government may unilaterally change its COR designation(s). ARTICLE G.3. KEY PERSONNEL The key personnel specified in this contract are considered to be essential to work performance. At least 30 days prior to diverting any of the specified individuals to other programs or contracts (or as soon as possible, if an individual must be replaced, for example, as a result of leaving the employ of the Contractor), the Contractor shall notify the Contracting Officer and shall submit comprehensive justification for the diversion or replacement request (including proposed substitutions for key personnel) to permit evaluation by the Government of the impact on performance under this contract. The Contractor shall not divert or otherwise replace any key personnel without the written consent of the Contracting Officer. The Government may modify the contract to add or delete key personnel at the request of the Contractor or Government. The following individuals are considered to be essential to the work being performed hereunder: Name Title [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] ARTICLE G.4. INVOICE SUBMISSION a. The Contractor shall submit an electronic copy of contract monthly invoices/financial reports to the Contracting Officer as defined above, in ARTICLE G of this contract. b. Contractor invoices/financial reports shall conform to the form, format, and content requirements of the instructions for Invoice/Financing requests made a part of the contract at Section J, Attachments 2 & 3. c. Monthly invoices must include the cumulative total expenses to date, adjusted (as applicable) to show any amounts suspended by the Government. d. The Contractor agrees to immediately notify the Contracting Officer in writing if there is an anticipated overrun (any amount) or unexpended balance (greater than 10 percent) of the estimated costs for the base period or any options for additional quantities (See estimated costs under Articles B.2 and B.3) and the reasons for the variance. Also refer to the requirements of FAR Clause 52.232-20, Limitation of Cost. e. The Contractor shall submit an electronic copy of the payment request to the approving official instead of a paper copy. The payment request shall be transmitted as an attachment via e-mail to the address listed above in one of the following formats: MSWord,

24 MS Excel, or Adobe Portable Document Format (PDF). Only one payment request shall be submitted per e-mail and the subject line of the e-mail shall include the Contractor's name, contract number, and unique invoice number. f. All invoice submissions shall be in accordance with FAR Clause 52.232-25, Prompt Payment. g. Invoices shall be delivered electronically to the Contracting Officer (CO), the Contracting Officer’s Representative (COR), PSC, and e-Room electronically. Unless otherwise specified by the Contracting Officer, all deliverables, invoices, and reports furnished to the Government under the resultant contract shall be addressed as follows: [**] Contracting Officer HHS/ASPR/AMCG 200 C Street, S.W. Washington, DC 20024 Email: [**] [**] Contracting Officer Representative HHS/ASPR/BARDA 200 C Street, S.W. Washington, DC 20024 Email: [**] Email invoices to: PSC_Invoices@psc.hhs.gov e-Room ARTICLE G.5. INDIRECT COST RATES The established provisional billing rates are based on rates approved by NIH-DFAS and adjustments made for consumable materials, which are specific to this contract HHSO100201600030C, per 2016 Provisional Billing Rate letter dated September 15, 2016. The following interim provisional indirect rates will be utilized for billing purposes during the period of performance: Fringe benefits at [**]%, Development O/H at [**]%, G&A at [**]% and Material and Subcontracting Handling at [**]%. Final rate proposals must be sent to the Contracting Officer upon immediate notification from the cognizant audit agency. See FAR Clause 52.216-7, Allowable Cost and Payment. ARTICLE G.6. PROVIDING ACCELERATED PAYMENT TO SMALL BUSINESS SUBCONTRACTORS, FAR 52.232-40 (December 2013) (a) Upon receipt of accelerated payments from the Government, the Contractor shall make accelerated payments to its small business subcontractors under this contract, to the maximum extent practicable and prior to when such payment is otherwise required under the applicable contract or subcontract, after receipt of a proper invoice and all other required documentation from the small business subcontractor. (b) The acceleration of payments under this clause does not provide any new rights under the prompt Payment Act. (c) Include the substance of this clause, include this paragraph c, in all subcontracts with small business concerns, including subcontracts with small business concerns for the acquisition of commercial items. ARTICLE G.7. POST AWARD EVALUATION OF CONTRACTOR PERFORMANCE 1. Contractor Performance Evaluations Interim and final evaluations of Contractor performance will be prepared on this contract in accordance with FAR Subpart 42.15. The final performance evaluation will be prepared at the time of completion of work. In addition to the final evaluation, interim evaluation(s) will be prepared annually as to coincide with the Anniversary date of the contract. Interim and final evaluations will be provided to the Contractor as soon as practicable after completion of the evaluation. The Contractor will be permitted thirty days to review the document and to submit additional information or a rebutting statement. If agreement cannot

25 be reached between the parties, the matter will be referred to an individual one level above the Contracting Officer whose decision will be final. Copies of the evaluations, Contractor responses, and review comments, if any, will be retained as part of the contract file, and may be used to support future award decisions. 2. Electronic Access to Contractor Performance Evaluations Contractors may access evaluations through a secure website for review and comment at the following: http://cpars.gov ARTICLE G.8. CONTRACT COMMUNICATIONS/CORRESPONDENCE The Contractor shall identify all correspondence, reports, and other data pertinent to this contract by imprinting the contract number HHSO100201600030C from Page 1 of the contract. ARTICLE G.9. GOVERNMENT PROPERTY 1. In addition to the requirements of the clause, GOVERNMENT PROPERTY, incorporated in SECTION I of this contract, the Contractor shall comply with the provisions of HHS Publication, "Contractor's Guide for Control of Government Property," which is incorporated into this contract by reference. This document can be accessed at: http://www.hhs.gov/hhsmanuals/ (HHS Logistics Management Manual) Among other issues, this publication provides a summary of the Contractor's responsibilities regarding purchasing authorizations and inventory and reporting requirements under the contract. 2. Notwithstanding the provisions outlined in the HHS Publication, "Contractor's Guide for Control of Government Property," which is incorporated in this contract in paragraph 1 above, the Contractor shall use the form entitled, "Report of Government Owned, Contractor Held Property" for submitting summary reports required under this contract, as directed by the Contracting Officer or his/her designee. This form is included as an attachment in SECTION J of this contract. 3. Title will vest in the Government for equipment purchased as a direct cost.

26 Section H – Special Contract Requirements ARTICLE H.1. PROTECTION OF HUMAN SUBJECTS a. The Contractor agrees that the rights and welfare of human subjects involved in research under this contract shall be protected in accordance with 45 CFR Part 46 and with the Contractor's current Assurance of Compliance on file with the Office for Human Research Protections (OHRP), Department of Health and Human Services. The Contractor further agrees to provide certification at least annually that the Institutional Review Board has reviewed and approved the procedures, which involve human subjects in accordance with 45 CFR Part 46 and the Assurance of Compliance. b. The Contractor shall bear full responsibility for the performance of all work and services involving the use of human subjects under this contract and shall ensure that work is conducted in a proper manner and as safely as is feasible. The parties hereto agree that the Contractor retains the right to control and direct the performance of all work under this contract. The Contractor shall not deem anything in this contract to constitute the Contractor or any subcontractor, agent or employee of the Contractor, or any other person, organization, institution, or group of any kind whatsoever, as the agent or employee of the Government. The Contractor agrees that it has entered into this contract and will discharge its obligations, duties, and undertakings and the work pursuant thereto, whether requiring professional judgment or otherwise, as an independent Contractor without imputing liability on the part of the Government for the acts of the Contractor or its employees. c. Contractors involving other agencies or institutions in activities considered to be engaged in research involving human subjects must ensure that such other agencies or institutions obtain their own FWA if they are routinely engaged in research involving human subjects or ensure that such agencies or institutions are covered by the Contractors’ FWA via designation as agents of the institution or via individual investigator agreements (see OHRP website at: http://www.hhs.gov/ohrp/policy/guidanceonalternativetofwa.pdf). d. If at any time during the performance of this contract, the Contracting Officer determines, in consultation with OHRP that the Contractor is not in compliance with any of the requirements and/or standards stated in paragraphs (a) and (b) above, the Contracting Officer may immediately suspend, in whole or in part, work and further payments under this contract until the Contractor corrects the noncompliance. The Contracting Officer may communicate the notice of suspension by telephone with confirmation in writing. If the Contractor fails to complete corrective action within the period of time designated in the Contracting Officer's written notice of suspension, the Contracting Officer may, after consultation with OHRP, terminate this contract in whole or in part, and the Contractor's name may be removed from the list of those Contractors with approved Human Subject Assurances. ARTICLE H.2. NON-CLINICAL RESEARCH PHS Policy on Humane Care and Use of Laboratory Animals Before initiation of research and then with the annual progress report, the Contractor must submit to the Government a copy of the current Institutional Animal Care and Use Committees (IACUC) documentation of continuing review and approval and the Office of Laboratory Animal Welfare (OLAW- National Institutes of Health) Federal Wide Assurance (FWA) number for the institution or site. If other institutions are involved in the research (e.g., a multicenter trial or study), each institution's IACUC must review and approve the protocol. They must also provide the Government initial documentation and documentation of continuing review and approval and FWA number.

27 The Contractor must ensure that the applications as well as all protocols are reviewed by the performing institution’s IACUC. To help ensure the safety of animals used in BARDA funded studies, the Contractor must provide the Government copies of documents related to all major changes in the status of ongoing protocols, including the following: a) All amendments or changes to the protocol, identified by protocol version number, date, or both and date it is valid. b) All material changes in IACUC policies and procedures, identified by version number, date, and all required signatories (if applicable) c) Termination or temporary suspension of the study(ies) for regulatory issues d) Termination or temporary suspension of the protocol. e) Any change that is made in the specific IACUC approval for the indicated study(ies). f) Any other problems or issues that could affect the scientific integrity of the study(ies), i.e. fraud, misrepresentation, misappropriation of funds, etc. Contractors must notify the Government by email of any of the above changes within three business days from the time Contractor becomes aware of such changes, followed by a letter signed by the institutional business official, detailing notification of the change of status to the local IACUC and a copy of any responses from the IACUC. If a non-clinical protocol has been reviewed by an institutional biosafety committee (IBC) or the NIH Recombinant DNA Advisory Committee (RAC), the Contractor must provide information about the initial and ongoing review and approval, if any. See the NIH Guidelines for Research Involving Recombinant DNA Molecules. Non-Clinical Data and Safety Monitoring Requirements The Contractor shall continue safety monitoring for all non-clinical studies of investigational drugs, devices, or biologics. FDA expects non-clinical studies to include safety in addition to efficacy. The Contractor should consider evaluation of clinical relevant safety markers in the pivotal and non-pivotal, non-clinical studies. BARDA will work with the Contractors on decisions regarding the type and extent of safety data accrual to be employed before the start of efficacy or safety studies. The Contractor shall inform the Government of any upcoming site visits and/or audits of CRO facilities funded under this effort. The Government reserves the right to accompany the Contractor on site visits and/or audits of CROs as the Government deems necessary. BARDA Review Process Before Non-Clinical Study Execution Begins The Government is under the same policy-driven assurances as NIH in that it has a responsibility to ensure that mechanisms and procedures are in place to protect the safety and welfare of animals used in BARDA funded non- clinical trials. Therefore, before study execution, the Contractor must provide the following (as applicable) for review and approval by the Government: 1. Non-clinical research protocol to be submitted for IACUC approval identified by version number, date, or both, including details of study design, euthanasia criteria, proposed interventions, and exclusion criteria. Contractor should reduce the number of animals required for a study using power of statistics 2. Plans for the management of side effects, rules for interventions and euthanasia criteria 3. Procedures for assessing and collecting safety data 4. If a study is contracted through CRO(s), work orders and service agreements the Contractor shall assure that an integrated safety documentation plan is in place for

28 the study site, pharmacy service records on the dosing material to be used and excipients, and laboratory services (including histopathology). 5. Documentation that the Contractor or CRO and all staff responsible for the conduct of the research have received required training in the protection and handling of animals 6. Purchasing of animals and/or other supplies for non-clinical studies funded in part or in whole by BARDA requires written approval by the Contracting Officer. The Contractor must have the ability to return/re-sell animals, at purchase price, to distributor or a third party, in the event that the protocols do not obtain approval 7. Provide justification for whether studies require good laboratory practice (GLP) conditions BARDA comments will be forwarded to the Contractor within two weeks (10 business days) of receipt of the above information. The Contractor must address in writing all study design, safety, regulatory, ethical, and conflict of interest concerns raised by the BARDA COR to the satisfaction of the Government before study execution. After the Government receives the corrected documentation, a written protocol approval will be provided by the COR to the Contractor. This written approval provides authorization to the Contractor to execute the specific non-clinical animal study funded in part or in whole by the Government. Documentation of IACUC approval, including OLAW FWA number, IACUC registration number, and IACUC name, must be provided to the BARDA COR within 24 hours of receipt by the Contractor. In case of problems or issues, the BARDA COR will contact the Contractor within two weeks (10 business days) by email or fax, followed within 30 calendar days by an official letter to the principal investigator, with a copy to the institution’s office of sponsored programs, listing issues and appropriate actions to be discussed. Final decisions regarding ongoing safety reporting requirements for research not performed under an Investigational New Drug Application (IND) must be made jointly by the Government and the Contractor. ARTICLE H.3. CLINICAL RESEARCH These Clinical Terms apply to all contracts that involve clinical research. The Government shall have unlimited rights to all protocols, data generated from the execution of these protocols, and final reports, funded by the Government under this contract, as defined in Rights in Data Clause in FAR 52.227-14. The Government reserves the right to request that the Contractor provide any contract deliverable in a non-proprietary form, to ensure the Government has the ability to review and distribute the deliverables, as the Government deems necessary. H.3.1 Safety and Monitoring Issues Institutional Review Board (IRB) or Independent Ethics Committee (IEC) Approval Before initiation of research and then with Annual Progress Reports, the Contractor shall submit to the Government a copy of the current IRB or IEC approved informed consent document, documentation of continuing review and approval and the Office of Human Research Protections (OHRP) FWA number for the institution or site. If other institutions are involved in the research (e.g., a multicenter clinical trial or study), each institution's IRB or IEC must review and approve the protocol. They must also provide the Government initial and annual documentation of continuing review and approval, including the current approved informed consent document and FWA number. The grantee institution must ensure that the applications as well as all protocols are reviewed by their IRB or IEC. To help ensure the safety of participants enrolled in BARDA-funded studies, the Contractor must provide the Government a summary explanation and copies of documents related to all major changes in the status of ongoing protocols, including the following:

29 1. All amendments or changes to the protocol, identified by protocol version number, date, or both and date it is valid. 2. All changes in informed consent documents, identified by version number, date, or both and dates it is valid. 3. Termination or temporary suspension of patient accrual. 4. Termination or temporary suspension of the protocol. 5. Any change in IRB approval. 6. Any other problems or issues that could affect the participants in the studies. Contractors must notify BARDA through the Contracting Officer’s Representative (COR) and Contracting Officer (CO) of any of the above changes within 24 hours from the time the Contractor becomes aware of the change by email, followed by a letter signed by the institutional business official, detailing notification of the change of status to the local IRB and a copy of any responses from the IRB or IEC. If a clinical protocol has been reviewed by an Institutional Bio-safety Committee (IBC) or the NIH Recombinant DNA Advisory Committee (RAC), the Contractor must provide information about the initial and ongoing review and approval, if any. See the NIH Guidelines for Research Involving Recombinant DNA Molecules. H.3.2. Data and Safety Monitoring Requirements The Contractor may be required to conduct independent safety monitoring for clinical trials of investigational drugs, devices, or biologics; clinical trials of licensed products; and clinical research of any type involving more than minimal risk to volunteers. Independent monitoring can take a variety of forms. Phase III clinical trials must have an assigned independent data and safety monitoring board (DSMB); other trials may require DSMB oversight as well. The Contractor shall inform the Government of any upcoming site visits and/or audits of Contractor facilities funded under this effort. BARDA reserves the right to accompany the Contractor on site visits and/or audits of Contractors and Subcontractors as the Government deems necessary. The type of monitoring to be used shall be mutually agreed upon between the Contractor and the Government before enrollment starts. Discussions with the responsible BARDA COR regarding appropriate safety monitoring and approval of the final monitoring plan by BARDA must occur before patient enrollment begins and may include discussions about the appointment of one of the following: 1. Independent Safety Monitor – a physician or other appropriate expert who is independent of the study and available in real time to review and recommend appropriate action regarding adverse events and other safety issues. 2. Independent Monitoring Committee (IMC) or Safety Monitoring Committee (SMC) – a small group of independent investigators and biostatisticians who review data from a particular study. 3. Data and Safety Monitoring Board – an independent committee charged with reviewing safety and trial progress and providing advice with respect to study continuation, modification, and termination. The Contractor may be required to use an established BARDA DSMB or to organize an independent DSMB. All phase III clinical trials must be reviewed by a DSMB; other trials may require DSMB oversight as well. Please refer to: NIAID Principles for Use of a Data and Safety Monitoring Board (DSMB) For Oversight of Clinical Trials Policy. The Government retains the right to place a nonvoting member on the DSMB. When a monitor or monitoring board is organized, a description of it, its charter or operating procedures (including a proposed meeting schedule and plan for review of adverse events), and roster and curriculum vitae from all members must be submitted to and approved by the Government before enrollment starts. Additionally, the Contractor must submit written summaries of all reviews conducted by the monitoring group to the Government within 30 days of reviews or meetings.

30 H.3.3. BARDA Protocol Review Process Before Patient Enrollment Begins BARDA has a responsibility to ensure that mechanisms and procedures are in place to protect the safety of participants in BARDA-supported clinical trials. Therefore, before patient accrual or participant enrollment, the Contractor must provide the following (as applicable) for review and approval by the Government: 1. Clinical research protocol to be submitted for approval by the IRB or IEC, identified by version number, date, or both, including details of study design, proposed interventions, patient eligibility, and exclusion criteria; 2. Informed consent document, identified by version number, date, or both and date it is valid; 3. Plans for the management of side effects; 4. Procedures for assessing and reporting adverse events; 5. Plans for data and safety monitoring (see B above) and monitoring of the clinical study site, pharmacy, and laboratory; 6. Documentation that the Contractor and all study staff responsible for the design or conduct of the research have received Good Clinical Practice (GCP) training in the protection of human subjects. BARDA comments will be forwarded to the Contractor within two weeks (10 business days) of receipt of the above information. The Contractor must address in writing all study design, safety, regulatory, ethical, and conflict of interest concerns raised by the BARDA COR to the satisfaction of the Government before patient accrual or participant enrollment can begin. After the Government receives the corrected documentation, a written protocol approval will be provided by the COR and CO to the Contractor. This written approval provides authorization to the Contractor to execute the specific clinical study funded in part or in whole by the Government. Documentation of IRB or IEC approval, including OHRP FWA number, IRB or IEC registration number, and IRB and IEC name, must be provided to the BARDA COR within 24 hours of receipt by the Contractor. H.3.4. Required Time-Sensitive Notification Under an IND or IDE, the sponsor must provide FDA safety reports of serious adverse events. Under these Clinical Terms of Award, the Contractor must submit copies to the responsible BARDA Contracting Officer's representative (COR) as follows: 1. Expedited safety report of unexpected or life-threatening experience or death – A copy of any report of unexpected or life-threatening experience or death associated with the use of an IND drug, which must be reported to FDA by telephone or fax as soon as possible but no later than seven days after the IND sponsor’s receipt of the information, must be submitted to the BARDA program officer or the Contracting Officer's Representative within 24 hours of FDA notification. 2. Expedited safety reports of serious and unexpected adverse experiences – A copy of any report of unexpected and serious adverse experience associated with use of an IND drug or any finding from tests in laboratory animals that suggests a significant risk for human subjects, which must be reported in writing to FDA as soon as possible but no later than 15 calendar days after the IND sponsor’s receipt of the information, must be submitted to the BARDA Contracting Officer’s Representative within 24 hours of FDA notification. 3. IDE reports of unanticipated adverse device effect – A copy of any reports of unanticipated adverse device effect submitted to FDA must be submitted to the BARDA Contracting Officer’s Representative within 24 hours of FDA notification. 4. Expedited safety reports – shall be sent to the BARDA COR concurrently with the report to FDA.

31 5. Other adverse events documented during the course of the trial shall be included in the annual IND report and reported to the BARDA annually. In case of problems or issues, the BARDA COR will contact the Contractor within 10 working days by email, followed within 7 calendar days by an official letter to the Contractor. The Contractor shall forward the official letter to the principal investigator listing issues and appropriate actions to be discussed. Safety reporting for research not performed under an IND. Ongoing safety reporting requirements for research not performed under an IND shall be mutually agreed upon by the BARDA Contracting Officer’s Representative and the Contractor. ARTICLE H.4. HUMAN MATERIALS The acquisition and supply of all human specimen material (including fetal material) used under this contract shall be obtained by the Contractor in full compliance with applicable State and Local laws and the provisions of the Uniform Anatomical Gift Act in the United States, and no undue inducements, monetary or otherwise, will be offered to any person to influence their donation of human material. ARTICLE H.5. CARE OF LIVE VERTEBRATE ANIMALS a. Before undertaking performance of any contract involving animal-related activities where the species is regulated by USDA, the Contractor shall register with the Secretary of Agriculture of the United States in accordance with 7 U.S.C. 2136 and 9 CFR sections 2.25 through 2.28. The Contractor shall furnish evidence of the registration to the Contracting Officer. b. The Contractor shall acquire vertebrate animals used in research from a dealer licensed by the Secretary of Agriculture under 7 U.S.C. 2133 and 9 CFR Sections 2.1- 2.11, or from a source that is exempt from licensing under those sections. c. The Contractor agrees that the care, use and intended use of any live vertebrate animals in the performance of this contract shall conform with the Public Health Service (PHS) Policy on Humane Care of Use of Laboratory Animals (PHS Policy), the current Animal Welfare Assurance (Assurance), the Guide for the Care and Use of Laboratory Animals (National Academy Press, Washington, DC) and the pertinent laws and regulations of the United States Department of Agriculture (see 7 U.S.C. 2131 et seq. and 9 CFR Subchapter A, Parts 1-4). In case of conflict between standards, the more stringent standard shall govern. d. If at any time during performance of this contract, the Contracting Officer determines, in consultation with the Office of Laboratory Animal Welfare (OLAW), National Institutes of Health (NIH), that the Contractor is not in compliance with any of the requirements and standards stated in paragraphs (a) through (c) above, the Contracting Officer may immediately suspend, in whole or in part, work and further payments under this contract until the Contractor corrects the noncompliance. Notice of the suspension may be communicated by telephone and confirmed in writing. If the Contractor fails to complete corrective action within the period of time designated in the Contracting Officer's written notice of suspension, the Contracting Officer may, in consultation with OLAW, NIH, terminate this contract in whole or in part, and the Contractor's name may be removed from the list of those contractors with approved Assurances. Note: The Contractor may request registration of its facility and a current listing of licensed dealers from the Regional Office of the Animal and Plant Health Inspection Service (APHIS), USDA, for the region in which its research facility is located. The location of the appropriate APHIS Regional Office, as well as information concerning this program may be obtained by contacting the Animal Care Staff, USDA/APHIS, 4700 River Road, Riverdale, Maryland 20737 (E-mail: ace@aphis.usda.gov; W eb site: (http://www.aphis.usda.gov/animal_welfare).

32 ARTICLE H.6. ANIMAL WELFARE All research involving live, vertebrate animals shall be conducted in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals. This policy may be accessed at:http://grants1.nih.gov/grants/olaw/references/phspol.htm ARTICLE H.7. INFORMATION ON COMPLIANCE WITH ANIMAL CARE REQUIREMENTS Registration with the U. S. Dept. of Agriculture (USDA) is required to use regulated species of animals for biomedical purposes. USDA is responsible for the enforcement of the Animal Welfare Act (7 U.S.C. 2131 et. seq.), http://www.nal.usda.gov/awic/legislat/awa.htm. The Public Health Service (PHS) Policy is administered by the Office of Laboratory Animal Welfare (OLAW) http://grants2.nih.gov/grants/olaw/olaw.htm. An essential requirement of the PHS Policy http://grants2.nih.gov/grants/olaw/references/phspol.htm is that every institution using live vertebrate animals must obtain an approved assurance from OLAW before they can receive funding from any component of the U. S. Public Health Service. The PHS Policy requires that Assured institutions base their programs of animal care and use on the Guide for the Care and Use of Laboratory Animals http://www.nap.edu/readingroom/books/labrats/ and that they comply with the regulations (9 CFR, Subchapter A)http://www.nal.usda.gov/awic/legislat/usdaleg1.htm issued by the U.S. Department of Agriculture (USDA) under the Animal Welfare Act. The Guide may differ from USDA regulations in some respects. Compliance with the USDA regulations is an absolute requirement of this Policy. The Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) http://www.aaalac.org is a professional organization that inspects and evaluates programs of animal care for institutions at their request. Those that meet the high standards are given the accredited status. As of the 2002 revision of the PHS Policy, the only accrediting body recognized by PHS is the AAALAC. While AAALAC Accreditation is not required to conduct biomedical research, it is highly desirable. AAALAC uses the Guide as their primary evaluation tool. They also use the Guide for the Care and Use of Agricultural Animals in Agricultural Research and Teaching. It is published by the Federated of Animal Science Societies http://www.fass.org. ARTICLE H.8. REQUIREMENTS FOR ADEQUATE ASSURANCE OF PROTECTION OF VERTEBRATE ANIMAL SUBJECTS The PHS Policy on Humane Care and Use of Laboratory Animals requires that applicant organizations proposing to use vertebrate animals file a written Animal Welfare Assurance with the Office for Laboratory Animal Welfare (OLAW), establishing appropriate policies and procedures to ensure the humane care and use of live vertebrate animals involved in research activities supported by the PHS. The PHS Policy stipulates that an applicant organization, whether domestic or foreign, bears responsibility for the humane care and use of animals in PHS- supported research activities. Also, the PHS policy defines “animal” as “any live, vertebrate animal used, or intended for use, in research, research training, experimentation, biological testing or for related purposes.” This Policy implements and supplements the U.S. Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training, and requires that institutions use the Guide for the Care and Use of Laboratory Animals as a basis for developing and implementing an institutional animal care and use program. This Policy does not affect applicable State or local laws or regulations that impose more stringent standards for the care and use of laboratory animals. All institutions are required to comply, as applicable, with the Animal Welfare Act as amended (7 USC 2131 et. seq.) and other Federal statutes and regulations relating to animals. These documents are available from the Office of Laboratory Animal Welfare, National Institutes of Health, Bethesda, MD 20892, (301) 496-7163. See http://grants.nih.gov/grants/olaw/olaw.htm. No PHS supported work for research involving vertebrate animals will be conducted by an organization, unless that organization is operating in accordance with an approved Animal

33 Welfare Assurance and provides verification that the Institutional Animal Care and Use Committee (IACUC) has reviewed and approved the proposed activity in accordance with the PHS policy. Applications may be referred by the PHS back to the institution for further review in the case of apparent or potential violations of the PHS Policy. No award to an individual will be made unless that individual is affiliated with an assured organization that accepts responsibility for compliance with the PHS Policy. Foreign applicant organizations applying for PHS awards for activities involving vertebrate animals are required to comply with PHS Policy or provide evidence that acceptable standards for the humane care and use of animals will be met. Foreign applicant organizations are not required to submit IACUC approval, but should provide information that is satisfactory to the Government to provide assurances for the humane care of such animals. ARTICLE H.9. APPROVAL OF REQUIRED ASSURANCE BY OLAW Under governing regulations, federal funds which are administered by the Department of Health and Human Services, Office of Biomedical Advanced Research and Development Authority (BARDA) shall not be expended by the Contractor for research involving live vertebrate animals, nor shall live vertebrate animals be involved in research activities by the Contractor under this award unless a satisfactory assurance of compliance with 7 U.S.C. 2316 and 9 CFR Sections 2.25-2.28 is submitted within 30 days of the initiation of research and approved by the Office of Laboratory Animal Welfare (OLAW). Each performance site (if any) must also assure compliance with 7 U.S.C. 2316 and 9 CFR Sections 2.25-2.28 with the following restriction: Only activities which do not directly involve live vertebrate animals (i.e. are clearly severable and independent from those activities that do involve live vertebrate animals) may be conducted by the Contractor or individual performance sites pending OLAW approval of their respective assurance of compliance with 7 U.S.C. 2316 and 9 CFR Sections 2.25-2.28. Additional information regarding OLAW may be obtained via the Internet at http://grants2.nih.gov/grants/olaw/references/phspol.htm ARTICLE H.10. NEEDLE EXCHANGE The Contractor shall not use contract funds to carry out any program of distributing sterile needles or syringes for the hypodermic injection of any illegal drug. ARTICLE H.11. ACKNOWLEDGEMENT OF FEDERAL FUNDING The Contractor shall clearly state, when issuing statements, press releases, requests for proposals, bid solicitations and other documents describing projects or programs funded in whole or in part with Federal money: (1) the percentage of the total costs of the program or project which will be financed with Federal money; (2) the dollar amount of Federal funds for the project or program; and (3) the percentage and dollar amount of the total costs of the project or program that will be financed by nongovernmental sources. ARTICLE H.12. CONTINUED BAN ON FUNDING ABORTION AND CONTINUED BAN OF FUNDING OF HUMAN EMBRYO RESEARCH a. The Contractor shall not use any funds obligated under this contract for any abortion. b. The Contractor shall not use any funds obligated under this contract for the following: 1. The creation of a human embryo or embryos for research purposes; or 2. Research in which a human embryo or embryos are destroyed, discarded, or knowingly subjected to risk of injury of death greater than that allowed for research on fetuses in utero under 45 CFR part 46 and Section 498(b) of the Public Health Service Act (42 U.S.C. 289g(b)). c. The term “human embryo or embryos'” includes any organism, not protected as a human subject under 45 CFR part 46 as of the date

34 of the enactment of this Act, that is derived by fertilization, parthenogenesis, cloning, or any other means from one or more human gametes of human diploid cells. d. The Contractor shall not use any Federal funds for the cloning of human beings. ARTICLE H.13. DISSEMINATION OF FALSE OR DELIBERATELY MISLEADING INFORMATION The Contractor shall not use contract funds to disseminate information that is deliberately false or misleading. ARTICLE H.14. OMB CLEARANCE In accordance with HHSAR 352.211-3, Paperwork Reduction Act, the Contractor shall not proceed with surveys or interviews until such time as Office of Management and Budget (OMB) Clearance for conducting interviews has been obtained by the Contracting Officer's Representative (COR) and the Contracting Officer has issued written approval to proceed. ARTICLE H.15. RESEARCH INVOLVING HUMAN FETAL TISSUE All research involving human fetal tissue shall be conducted in accordance with the Public Health Service Act, 42 U.S.C. 289g-1 and 289g-2. Implementing regulations and guidance for conducting research on human fetal tissue may be found at 45 CFR 46, Subpart B and http://grants1.nih.gov/grants/guide/notice-files/not93-235.html and any subsequent revisions to this NIH Guide to Grants and Contracts ("Guide") Notice. The Contractor shall make available, for audit by the Secretary, HHS, the physician statements and informed consents required by 42 USC 289g-1(b) and (c), or ensure HHS access to those records, if maintained by an entity other than the Contractor. ARTICLE H.16. REPORTING MATTERS INVOLVING FRAUD, WASTE, AND ABUSE Anyone who becomes aware of the existence or apparent existence of fraud, waste and abuse in BARDA funded programs is encouraged to report such matters to the HHS Inspector General's Office in writing or on the Inspector General's Hotline. The toll free number is 1-800-HHS-TIPS (1-800-447-8477). All telephone calls will be handled confidentially. The e-mail address is Htips@os.dhhs.gov and the mailing address is: Office of Inspector General Department of Health and Human Services TIPS HOTLINE P.O. Box 23489 Washington, D.C. 20026 ARTICLE H.17. PROHIBITION ON CONTRACTOR INVOLVEMENT WITH TERRORIST ACTIVITIES The Contractor acknowledges that U.S. Executive Orders and Laws, including but not limited to E.O. 13224 and P.L. 107-56, prohibit transactions with, and the provision of resources and support to, individuals and organizations associated with terrorism. It is the legal responsibility of the Contractor to ensure compliance with these Executive Orders and Laws. This clause must be included in all subcontracts issued under this contract. ARTICLE H.18. RESTRICTION ON PORNOGRAPHY ON COMPUTER NETWORKS The Contractor shall not use contract funds to maintain or establish a computer network unless such network blocks the viewing, downloading, and exchanging of pornography. ARTICLE H.19. CERTIFICATION OF FILING AND PAYMENT OF TAXES The Contractor must be in compliance with Section 518 of the Consolidated Appropriations Act of FY 2014.

35 ARTICLE H.20. ELECTRONIC INFORMATION AND TECHNOLOGY ACCESSIBILITY NOTICE a. Section 508 of the Rehabilitation Act of 1973 (29 U.S.C. 794d), as amended by the Workforce Investment Act of 1998 and the Architectural and Transportation Barriers Compliance Board Electronic and Information (EIT) Accessibility Standards (36 CFR part 1194), require that when Federal agencies develop, procure, maintain, or use electronic and information technology, Federal employees with disabilities have access to and use of information and data that is comparable to the access and use by Federal employees who are not individuals with disabilities, unless an undue burden would be imposed on the agency. Section 508 also requires that individuals with disabilities, who are members of the public seeking information or services from a Federal agency, have access to and use of information and data that is comparable to that provided to the public who are not individuals with disabilities, unless an undue burden would be imposed on the agency. b. Accordingly, any Offeror responding to this solicitation must comply with established HHS EIT accessibility standards. Information about Section 508 is available at http://www.hhs.gov/web/508. The complete text of the Section 508 Final Provisions can be accessed at http://www.access-board.gov/sec508/standards.htm. c. The Section 508 accessibility standards applicable to this solicitation are stated in the clause at 352.239-74, Electronic and Information Technology Accessibility. In order to facilitate the Government's determination whether proposed EIT supplies meet applicable Section 508 accessibility standards, Offerors must submit an HHS Section 508 Product Assessment Template, in accordance with its completion instructions. The purpose of the template is to assist HHS acquisition and program officials in determining whether proposed EIT supplies conform to applicable Section 508 accessibility standards. The template allows Offerors or developers to self-evaluate their supplies and document--in detail-- whether they conform to a specific Section 508 accessibility standard, and any underway remediation efforts addressing conformance issues. Instructions for preparing the HHS Section 508 Evaluation Template are available under Section 508 policy on the HHS Web site http://hhs.gov/web/508. In order to facilitate the Government's determination whether proposed EIT services meet applicable Section 508 accessibility standards, Offerors must provide enough information to assist the Government in determining that the EIT services conform to Section 508 accessibility standards, including any underway remediation efforts addressing conformance issues. d. Respondents to this solicitation must identify any exception to Section 508 requirements. If a Offeror claims its supplies or services meet applicable Section 508 accessibility standards, and it is later determined by the Government, i.e., after award of a contract or order, that supplies or services delivered do not conform to the described accessibility standards, remediation of the supplies or services to the level of conformance specified in the contract will be the responsibility of the Contractor at its expense. (End of provision) ARTICLE H.21. FULL EARNED VALUE MANAGEMENT SYSTEM a. The Contractor shall use an Earned Value Management System (EVMS) that is compliant with the guidelines in ANSI/EIA Standard-748 (current version at the time of award) to manage this contract. If the Contractor's current EVMS is not compliant at the time of award, see paragraph (b) of this clause. The Contractor shall submit EVM reports in accordance with the requirements of this contract. b. If, at the time of award, the Contractor's EVM system is not in compliance with the EVMS guidelines in ANSI/EIA Standard-748 (current version at time of award), the Contractor shall:

36 a. Apply the current system to the contract; and b. Take necessary and timely actions to meet the milestones in the Contractor's EVMS plan approved by the Contracting Officer. c. HHS will not formally validate or accept the Contractor's EVMS with respect to this contract. The use of the Contractor's EVMS for this contract does not imply HHS acceptance of the Contractor's EVMS for application to future contracts. The Contracting Officer or designee will conduct a Compliance Review to assess the Contractor's compliance with its approved plan. If the Contractor does not follow the approved implementation schedule or correct all resulting system deficiencies noted during the Compliance Review within a reasonable time, the Contracting Officer may take remedial action that may include, but is not limited to, suspension of or reduction in progress payments, or a reduction in fee. d. HHS will conduct a Performance Measurement Baseline Review (PMBR). If a pre-award PMBR has not been conducted, a post-award PMBR will be conducted by HHS as early as practicable, but no later than ninety (90) days after contract award. The Contracting Officer may also require a PMBR as part of the exercise of an option or the incorporation of a major modification. e. Unless a waiver is granted by the CFA, Contractor-proposed EVMS changes require approval of the CFA prior to implementation. The CFA will advise the Contractor of the acceptability of such changes within 30 calendar days after receipt of the notice of proposed changes from the Contractor. If the advance approval requirements are waived by the CFA, the Contractor shall disclose EVMS changes to the CFA at least 14 calendar days prior to the effective date of implementation. f. Unless a waiver is granted by the CFA, Contractor-proposed EVMS changes require approval of the CFA prior to implementation. The CFA will advise the Contractor of the acceptability of such changes within 30 calendar days after receipt of the notice of proposed changes from the Contractor. If the advance approval requirements are waived by the CFA, the Contractor shall disclose EVMS changes to the CFA at least 14 calendar days prior to the effective date of implementation. g. The Contractor shall provide access to all pertinent records and data requested by the Contracting Officer or a duly authorized representative as necessary to permit Government surveillance to ensure that the EVMS conforms, and continues to conform to the requirements referenced in paragraph (a) of this clause. ARTICLE H.22. CONFIDENTIALITY OF INFORMATION a. Confidential information, as used in this article, means information or data of a personal nature about an individual, or proprietary information or data submitted by or pertaining to an institution or organization. b. The Contracting Officer and the Contractor may, by mutual consent, identify elsewhere in this contract specific information and/or categories of information which the Government will furnish to the Contractor or that the Contractor is expected to generate which is confidential. Similarly, the Contracting Officer and the Contractor may, by mutual consent, identify such confidential information from time to time during the performance of the contract. Failure to agree will be settled pursuant to the "Disputes" clause. c. If it is established elsewhere in this contract that information to be utilized under this contract, or a portion thereof, is subject to the Privacy Act, the Contractor will follow the rules and procedures of disclosure set forth in the Privacy Act of 1974, 5 U.S.C. 552a, and implementing regulations and policies, with respect to systems of records determined to be subject to the Privacy Act. d. Confidential information, as defined in paragraph (a) of this article, shall not be disclosed without the prior written consent of the individual, institution, or organization.

37 e. Whenever the Contractor is uncertain with regard to the proper handling of material under the contract, or if the material in question is subject to the Privacy Act or is confidential information subject to the provisions of this article, the Contractor shall obtain a written determination from the Contracting Officer prior to any release, disclosure, dissemination, or publication. f. Contracting Officer determinations will reflect the result of internal coordination with appropriate program and legal officials. g. The provisions of paragraph (d) of this article shall not apply to conflicting or overlapping provisions in other Federal, State or local laws. ARTICLE H.23. INSTITUTIONAL RESPONSIBILITY REGARDING INVESTIGATOR FINANCIAL CONFLICTS OF INTERESTS The Institution (includes any Contractor, public or private, excluding a Federal agency) shall comply with the requirements of 45 CFR Part 94, Responsible Prospective Contractors, which promotes objectivity in research by establishing standards to ensure that Investigators (defined as the project director or principal Investigator and any other person, regardless of title or position, who is responsible for the design, conduct, or reporting of research funded under BARDA contracts, or proposed for such funding, which may include, for example, collaborators or consultants) will not be biased by any Investigator financial conflicts of interest. 45 CFR Part 94 is available at the following Web site: http://www.ecfr.gov/cgi-bin/text- idx?c=ecfr&SID=0af84ca649a74846f102aaf664da1623&rgn=div5&view=text&node=45:1.0.1.1.51 &idno=45 As required by 45 CFR Part 94, the Institution shall, at a minimum: a. Maintain an up-to-date, written, enforceable policy on financial conflicts of interest that complies with 45 CFR Part 94, inform each Investigator of the policy, the Investigator's reporting responsibilities regarding disclosure of significant financial interests, and the applicable regulation, and make such policy available via a publicly accessible Web site, or if none currently exist, available to any requestor within five business days of a request. A significant financial interest means a financial interest consisting of one or more of the following interests of the Investigator (and those of the Investigator's spouse and dependent children) that reasonably appears to be related to the Investigator's institutional responsibilities: 1. With regard to any publicly traded entity, a significant financial interest exists if the value of any remuneration received from the entity in the twelve months preceding the disclosure and the value of any equity interest in the entity as of the date of disclosure, when aggregated, exceeds $5,000. Included are payments and equity interests; 2. With regard to any non-publicly traded entity, a significant financial interest exists if the value of any remuneration received from the entity in the twelve months preceding the disclosure, when aggregated, exceeds $5,000, or when the Investigator (or the Investigator's spouse or dependent children) holds any equity interest; or 3. Intellectual property rights and interests, upon receipt of income related to such rights and interest. Significant financial interests do not include the following: 1. Income from seminars, lectures, or teaching, and service on advisory or review panels for government agencies, Institutions of higher education, academic teaching hospitals, medical centers, or research institutes with an Institution of higher learning; and 2. Income from investment vehicles, such as mutual funds and retirement accounts, as long as the Investigator does not directly control the investment decisions

38 made in these vehicles. b. Require each Investigator to complete training regarding the Institution's financial conflicts of interest policy prior to engaging in research related to any BARDA funded contract and at least every four years. The Institution must take reasonable steps [see Part 94.4(c)] to ensure that investigators working as collaborators, consultants or subcontractors comply with the regulations. c. Designate an official(s) to solicit and review disclosures of significant financial interests from each Investigator who is planning to participate in, or is participating in, the BARDA funded research. d. Require that each Investigator who is planning to participate in the BARDA funded research disclose to the Institution's designated official(s) the Investigator's significant financial interest (and those of the Investigator's spouse and dependent children) no later than the date of submission of the Institution's proposal for BARDA funded research. Require that each Investigator who is participating in the BARDA funded research to submit an updated disclosure of significant financial interests at least annually, in accordance with the specific time period prescribed by the Institution during the period of the award as well as within thirty days of discovering or acquiring a new significant financial interest. e. Provide guidelines consistent with the regulations for the designated official(s) to determine whether an Investigator's significant financial interest is related to BARDA funded research and, if so related, whether the significant financial interest is a financial conflict of interest. An Investigator's significant financial interest is related to BARDA funded research when the Institution, thorough its designated official(s), reasonably determines that the significant financial interest: Could be affected by the BARDA funded research; or is in an entity whose financial interest could be affected by the research. A financial conflict of interest exists when the Institution, through its designated official(s), reasonably determines that the significant financial interest could directly and significantly affect the design, conduct, or reporting of the BARDA funded research. f. Take such actions as necessary to manage financial conflicts of interest, including any financial conflicts of a subcontractor Investigator. Management of an identified financial conflict of interest requires development and implementation of a management plan and, if necessary, a retrospective review and mitigation report pursuant to Part 94.5(a). g. Provide initial and ongoing FCOI reports to the Contracting Officer pursuant to Part 94.5(b). h. Maintain records relating to all Investigator disclosures of financial interests and the Institution's review of, and response to, such disclosures, and all actions under the Institution's policy or retrospective review, if applicable, for at least 3 years from the date of final payment or, where applicable, for the other time periods specified in 48 CFR Part 4, subpart 4.7, Contract Records Retention. i. Establish adequate enforcement mechanisms and provide for employee sanctions or other administrative actions to ensure Investigator compliance as appropriate. j. Complete the certification in Section K - Representations, Certifications, and Other Statements of Contractors titled "Certification of Institutional Policy on Financial Conflicts of Interest". If the failure of an Institution to comply with an Institution's financial conflicts of interest policy or a financial conflict of interest management plan appears to have biased the design, conduct, or reporting of the BARDA funded research, the Institution must promptly notify the Contracting Officer of the corrective action taken or to be taken. The Contracting Officer will consider the situation and, as necessary, take appropriate action or refer the matter to the Institution for further action, which may include directions to the Institution on how to maintain appropriate objectivity in the BARDA funded research project.

39 The Contracting Officer and/or HHS may inquire at any time before, during, or after award into any Investigator disclosure of financial interests, and the Institution's review of, and response to, such disclosure, regardless of whether the disclosure resulted in the Institution's determination of a financial conflict of interests. The Contracting Officer may require submission of the records or review them on site. On the basis of this review of records or other information that may be available, the Contracting Officer may decide that a particular financial conflict of interest will bias the objectivity of the BARDA funded research to such an extent that further corrective action is needed or that the Institution has not managed the financial conflict of interest in accordance with Part 94.6(b). The issuance of a Stop Work Order by the Contracting Officer may be necessary until the matter is resolved. If the Contracting Officer determines that BARDA funded clinical research, whose purpose is to evaluate the safety or effectiveness of a drug, medical device, or treatment, has been designed, conducted, or reported by an Investigator with a financial conflict of interest that was not managed or reported by the Institution, the Institution shall require the Investigator involved to disclose the financial conflict of interest in each public presentation of the results of the research and to request an addendum to previously published presentations. ARTICLE H.24. PUBLICATION AND PUBLICITY The Contractor shall acknowledge the support of the Department of Health and Human Services, Office of the Assistant Secretary for Preparedness and Response, Biomedical Advanced Research and Development Authority whenever publicizing the work under this contract in any media by including an acknowledgment substantially as follows: "This project has been funded in whole or in part with Federal funds from the Office of the Assistant Secretary for Preparedness and Response, Biomedical Advanced Research and Development Authority, under Contract No. HHSO100201600030C" Press releases shall be considered to include the public release of information to any medium, excluding peer-reviewed scientific publications. The Contractor shall ensure that the Contracting Officer's Representative (COR) has received an advance copy of any press release related to this contract not less than five (5) working days prior to the issuance of the press release. ARTICLE H.25. ACCESS TO DOCUMENTATION/DATA The Government shall have physical and electronic access to all documentation and data generated under this contract, including: all data documenting Contractor performance, all data generated, all communications and correspondence with regulatory agencies and bodies to include all audit observations, inspection reports, milestone completion documents, and all Contractor commitments and responses. Contractor shall provide the Government with an electronic copy of all correspondence with the FDA, within 5 business days of receipt. The Government shall acquire unlimited rights to all data funded under a contract awarded in response to this RFP in accordance with FAR Subpart 27.4 and FAR Clause 52.227-14. ARTICLE H.26. DISSEMINATION OF INFORMATION No information related to data obtained under this contract shall be released or publicized without the prior written consent of the COR, whose approval shall not be unreasonably withheld, conditioned, or delayed, provided that no such consent is required to comply with any law, rule, regulation, court ruling or similar order; for submission to any government entity’ for submission to any securities exchange on which the Contractor’s (or its parent corporation’s) securities may be listed for trading; or to third parties relating to securing, seeking, establishing or maintaining regulatory or other legal approvals or compliance, financing and capital raising activities, or mergers, acquisitions, or other business transactions.

40 ARTICLE H.27. DISSEMINATION OF FALSE OR DELIBERATELY MISLEADING INFORMATION The Contractor shall not use contract funds to disseminate information that is deliberately false or misleading. ARTICLE H.28. IDENTIFICATION AND DISPOSITION OF DATA The Contractor will be required to provide certain data generated under this contract to the Department of Health and Human Services (HHS). HHS reserves the right to review any other data determined by HHS to be relevant to this contract. The Contractor shall keep copies of all data required by the Food and Drug Administration (FDA) relevant to this contract for the time specified by the FDA. ARTICLE H.29. CONFLICT OF INTEREST The Contractor represents and warrants that, to the best of the Contractor's knowledge and belief, there are no relevant facts or circumstances which could give rise to an organizational conflict of interest, as defined in FAR 2.101 and Subpart 9.5, or that the Contractor has disclosed all such relevant information. Prior to commencement of any work, the Contractor agrees to notify the Contracting Officer promptly that, to the best of its knowledge and belief, no actual or potential conflict of interest exists or to identify to the Contracting Officer any actual or potential conflict of interest the firm may have. In emergency situations, however, work may begin but notification shall be made within five (5) working days. The Contractor agrees that if an actual or potential organizational conflict of interest is identified during performance, the Contractor shall promptly make a full disclosure in writing to the Contracting Officer. This disclosure shall include a description of actions which the Contractor has taken or proposes to take, after consultation with the Contracting Officer, to avoid, mitigate, or neutralize the actual or potential conflict of interest. The Contractor shall continue performance until notified by the Contracting Officer of any contrary action to be taken. Remedies include termination of this contract for convenience, in whole or in part, if the Contracting Officer deems such termination necessary to avoid an organizational conflict of interest. If the Contractor was aware of a potential organizational conflict of interest prior to award or discovered an actual or potential conflict after award and did not disclose it or misrepresented relevant information to the Contracting Officer, the Government may terminate the contract for default, debar the Contractor from Government contracting, or pursue such other remedies as may be permitted by law or this contract. ARTICLE H.30. IN-PROCESS REVIEW In Process Reviews (IPR) will be conducted at the discretion of the Government to discuss the progression of the milestones. The Government reserves the right to revise the milestones and budget pending the development of the project. Deliverables may be required when the IPRs are conducted. The Contractor’s success in completing the required tasks under each work segment must be demonstrated through the Deliverables and Milestones specified under SECTION F. Those deliverables will constitute the basis for the Government’s decision, at its sole discretion, to proceed with the work segment, or unilaterally institute changes to the work segment, or terminate the work segment. IPRs may be scheduled at the discretion of the Government to discuss progression of the contract. The Contractor shall provide a presentation following a prescribed template which will be provided by the Government at least 30 days prior to the IPR. The Contractor shall provide a draft presentation to the Contracting Officer at least 10 days prior to the IPR. ARTICLE H.31. PRIVACY ACT APPLICABILITY 1) Notification is hereby given that the Contractor and its employees are subject to criminal penalties for violation of the Privacy Act to the same extent as employees of the Government. The Contractor shall assure that each of its employees knows the prescribed rules of conduct and that each is aware that he or she can be subjected to criminal penalty for violation of the Act. A copy of 45 CFR Part 5b, Privacy Act Regulations, may be obtained at http://www.gpoaccess.gov/cfr/index.html

41 2) The Project Officer/COR is hereby designated as the official who is responsible for monitoring Contractor compliance with the Privacy Act. 3) The Contractor shall follow the Privacy Act guidance as contained in the Privacy Act System of Records number 09-25-0200. This document may be obtained at the following link: http://oma.od.nih.gov/ms/privacy/pa- files/0200.htm ARTICLE H.32. QA AUDIT REPORTS BARDA reserves the right to participate in QA audits. Upon completion of the audit/site visit the Contractor shall provide a report capturing the findings, results and next steps in proceeding with the subcontractor. If action is requested of the subcontractor, detailed concerns for addressing areas of non-conformance to FDA regulations for GLP, GMP, or GCP guidelines, as identified in the audit report, must be provided to BARDA. The Contractor shall provide responses from the subcontractors to address these concerns and plans for corrective action execution. • Contractor shall notify CO and COR of upcoming, ongoing, or recent audits/site visits of subcontractors as part of weekly communications. The Contractor shall notify the CO and COR reasonably in advance of upcoming QA audit so that Government personnel may participate in person at BARDA’s discretion. • Contractor shall notify the COR and CO within 5 business days of report completion. ARTICLE H.33. BARDA AUDITS Contractor shall accommodate periodic or ad hoc site visits by the Government. If the Government, the Contractor, or other parties identifies any issues during an audit, the Contractor shall capture the issues, identify potential solutions, and provide a report to the Government. • If issues are identified during the audit, Contractor shall submit a report to the CO and COR detailing the finding and corrective action(s) within 10 business days of the audit. • COR and CO will review the report and provide a response to the Contractor with 10 business days. • Once corrective action is completed, the Contractor will provide a final report to the CO and COR. ARTICLE H.34. SECURITY REPORTING REQUIREMENT Violations of established security protocols shall be reported to the CO and COR upon discovery within 24 hours of its receipt of any compromise, intrusion, loss or interference of its security processes and procedures. The Contractor shall ensure that all software components that are not required for the operation and maintenance of the database/control system has been removed and/or disabled. The Contractor shall provide to the CO and the COR information appropriate to Information and Information Technology software and service updates and/or workarounds to mitigate all vulnerabilities associated with the data and shall maintain the required level of system security. The Contractor will investigate violations to determine the cause, extent, loss or compromise of sensitive program information, and corrective actions taken to prevent future violations. The CO in coordination with BARDA will determine the severity of the violation. Any contractual actions resulting from the violation will be determined by the CO.

42 PART II - CONTRACT CLAUSES SECTION I - CONTRACT CLAUSES ARTICLE I.1. FAR 52.252-2, CLAUSES INCORPORATED BY REFERENCE (FEBRUARY 1998) This contract incorporates the following clauses by reference, with the same force and effect as if they were given in full text. Upon request, the Contracting Officer will make their full text available. Also, the full text of a clause may be accessed electronically at these addresses: https://www.acquisition.gov/FAR/ . HHSAR Clauses at: http://www.hhs.gov/policies/hhsar/subpart352.html . General Clauses for Cost-Reimbursement/Fixed Price Research and Development Contract (1) FEDERAL ACQUISITION REGULATION (FAR) (48 CFR CHAPTER 1) CLAUSES: Reg Clause Date Clause Title FAR 52.202-1 Nov 2013 Definitions FAR 52.203-3 Apr 1984 Gratuities FAR 52.203-5 May 2014 Covenant Against Contingent Fees FAR 52.203-6 Sep 2006 Restrictions on Subcontractor Sales to the Government FAR 52.203-7 May 2014 Anti-Kickback Procedures FAR 52.203-8 May 2014 Cancellation, Rescission, and Recovery of Funds for Illegal or Improper Activity FAR 52.203-10 May 2014 Price or Fee Adjustment for Illegal or Improper Activity FAR 52.203-12 Oct 2010 Limitation on Payments to Influence Certain Federal Transactions FAR 52.203-13 Oct 2015 Contractor Code of Business Ethics and Conduct FAR 52.203-14 Oct 2015 Display of Hotline Poster(s) FAR 52.203-17 Apr 2014 Contractor Employee Whistleblower Rights and Requirement To Inform Employees of Whistleblower Rights FAR 52.204-4 May 2011 Printed or Copied Double-Sided on Postconsumer Fiber Content Paper FAR 52.204-7 Jul 2013 System for Award Management FAR 52.204-10 Oct 2015 Reporting Executive Compensation and First-Tier Subcontract Awards FAR 52.204-13 Jul 2013 System for Award Management Maintenance FAR 52.209-6 Oct 2015 Protecting the Government's Interests When Subcontracting With Contractors Debarred, Suspended, or Proposed for Debarment FAR 52.209-10 Nov 2015 Prohibition on Contracting with Inverted Domestic Corporations FAR 52.210-1 Apr 2011 Market Research FAR 52.215-2 Oct 2010 Audit and Records – Negotiation FAR 52.215-8 Oct 1997 Order of Precedence - Uniform Contract Format FAR 52.215-10 Aug 2011 Price Reduction for Defective Cost or Pricing Data FAR 52.215-11 Aug 2011 Price Reduction for Defective Certified Cost or Pricing Data—Modifications. FAR 52.215-12 Oct 2010 Subcontractor Certified Cost or Pricing Data FAR 52.215-13 Oct 2010 Subcontractor Certified Cost or Pricing Data—Modifications FAR 52.215-15 Oct 2010 Pension Adjustments and Asset Reversions FAR 52.215-17 Oct 1997 Waiver of Facilities Capital Cost of Money FAR 52.215-18 Jul 2005 Reversion or Adjustment of Plans for Postretirement Benefits (PRB) other than Pensions FAR 52.215-19 Oct 1997 Notification of Ownership Changes FAR 52.215-21 Oct 2010 Requirements for Certified Cost or Pricing Data and Data Other Than Certified Cost or Pricing Data -Modifications FAR 52.215-23 Oct 2009 Limitations on Pass-Through Charges FAR 52.216-7 Jun 2013 Allowable Cost and Payment FAR 52.216-8 Jun 2011 Fixed Fee FAR 52.219-8 Oct 2014 Utilization of Small Business Concerns FAR 52.219-28 July 2013 Post-Award Small Business Program Representation FAR 52.222-1 Feb 1997 Notice to the Government of Labor Disputes FAR 52.222-2 Jul 1990 Payment for Overtime Premiums

43 FAR 52.222-3 Jun2003 Convict Labor FAR 52.222-21 Apr 2015 Prohibition of Segregated Facilities FAR 52.222-26 Apr 2015 Equal Opportunity FAR 52.222-35 Oct 2015 Equal Opportunity for Veterans FAR 52.222-36 Jul 2014 Equal Opportunity for Workers with Disabilities FAR 52.222-37 Feb 2016 Employment Reports on Veterans FAR 52.222-40 Dec 2010 Notification of Employee Rights Under the National Labor Relations Act FAR 52.222-43 May 2014 Fair Labor Standards Act and Service Contract Labor Standards—Price Adjustment (Multiple Year and Option Contracts) FAR 52.222-50 Mar 2015 Combating Trafficking in Persons FAR 52.222-54 Oct 2015 Employment Eligibility Verification FAR 52.223-6 May 2001 Drug-Free Workplace FAR 52.223-18 Aug 2011 Encouraging Contractor Policy to Ban Text Messaging While Driving FAR 52.224-1 April 1984 Privacy Act Notification FAR 52.224-2 April 1984 Privacy Act FAR 52.225-13 Jun 2008 Restrictions on Certain Foreign Purchases FAR 52.227-1 Dec 2007 Authorization and Consent, Alternate 1 (APR 1984) FAR 52.227-2 Dec 2007 Notice and Assistance Regarding Patent and Copyright Infringement FAR 52.227-3 Apr 1984 Patent Indemnity FAR 52.227-11 May 2014 Patent Rights – Ownership by the Contractor FAR 52.227-14 May 2014 Rights in Data - General FAR 52.227-16 Jun 1987 Additional Data Requirements FAR 52.228-7 Mar 1996 Insurance – Liability to Third Persons FAR 52.229-3 Feb 2013 Federal, State and Local Taxes FAR 52.230-2 Oct 2015 Cost Accounting Standards FAR 52.230-6 June 2010 Administration of Cost Accounting Standards FAR 52.232-1 Apr 1984 Payments FAR 52.232-2 Apr 1984 Payments under Fixed-Price Research and Development Contracts FAR 52.232-8 Feb 2002 Discounts for Prompt Payment FAR 52.232-9 Apr 1984 Limitation on Withholding of Payments FAR 52.232-11 Apr 1984 Extras FAR 52.232-17 May 2014 Interest FAR 52.232-20 Apr 1984 Limitation of Cost FAR 52.232-23 May 2014 Assignment of Claims FAR 52.232-25 Jul 2013 Prompt Payment FAR 52.232-33 Jul 2013 Payment by Electronic Funds Transfer--System for Award Management FAR 52.233-1 May 2014 Disputes FAR 52.233-3 Aug 1996 Protest After Award, Alternate I FAR 52.233-4 Oct 2004 Applicable Law for Breach of Contract Claim FAR 52.242-1 Apr 1984 Notice of Intent to Disallow Costs FAR 52.242-3 May 2014 Penalties for Unallowable Costs FAR 52.242-4 Jan 1997 Certification of Final Indirect Costs FAR 52.242-13 Jul 1995 Bankruptcy FAR 52.243-1 Aug 1987 Changes - Fixed-Price Alternate V (Apr 1984). FAR 52.243-2 Aug 1987 Changes—Cost-Reimbursement Alternate V (Apr 1984). FAR 52.243.6 Apr 1984 Change Order Accounting FAR 52.243-7 Apr 1984 Notification of Changes FAR 52.244-2 Oct 2010 Subcontracts, Alternate 1 (Jun 2007) FAR 52.244-5 Dec 1996 Competition in Subcontracting FAR 52.244-6 Apr 2015 Subcontracts for Commercial Items FAR 52.245-1 Apr 2012 Government Property FAR 52.245-9 Apr 2012 Use and Charges FAR 52.246-7 Apr 1996 Inspection of Research and Development – Fixed-Price FAR 52.246-8 May 2001 Inspection of Research and Development – Cost-Reimbursement FAR 52.246-23 Feb 1997 Limitation of Liability. FAR 52.246-25 Feb 1997 Limitation of Liability—Services FAR 52.248-1 Oct 2010 Value Engineering

44 FAR 52.249-2 Apr 2012 Termination for the Convenience of the Government (Fixed-Price) FAR 52.249-6 May 2004 Termination (Cost-Reimbursement) FAR 52.249-8 Apr 1984 Default (Fixed-Price Supply and Service) FAR 52.249-9 Apr 1984 Default (Fixed-Price Research and Development) FAR 52.249-14 Apr 1984 Excusable Delays FAR 52.253-1 Jan 1991 Computer Generated Forms (2) DEPARTMENT OF HEALTH AND HUMAN SERVICES ACQUISITION REGULATION (HHSAR) (48 CFR CHAPTER 3) CLAUSES: HHSAR 352.203-70 Dec 2015 Anti-Lobbying HHSAR 352.211-3 Dec 2015 Paperwork Reduction Act HHSAR 352.222-70 Dec 2015 Contractor Cooperation in Equal Employment Opportunity Investigations HHSAR 352.223-70 Dec 2015 Safety and Health HHSAR 352.224-70 Dec 2015 Privacy Act HHSAR 352.227-70 Dec 2015 Publications and Publicity HHSAR 352.233-71 Dec 2015 Litigation and Claims HHSAR 352.237-75 Dec 2015 Key Personnel HHSAR 352.270-4a Dec 2015 Protection of Human Subjects HHSAR 352.270-5b Dec 2015 Care of Live Vertebrate Animals HHSAR 352.270-6 Dec 2015 Restriction on use of Human Subjects ARTICLE I.2. ADDITIONAL FAR CLAUSES INCLUDED IN FULL TEXT FAR 52.217-7 Option for Increased Quantity-Separately Priced Line Item (Mar 1989) The Government may require the delivery of the numbered line item, identified in the Schedule as an option item, in the quantity and at the price stated in the Schedule. The Contracting Officer may exercise the option by written notice to the Contractor within 30 days. Delivery of added items shall continue at the same rate that like items are called for under the contract, unless the parties otherwise agree. FAR 52.217-9 Option to Extend the Term of the Contract (Mar 2000) (a) The Government may extend the term of this contract by written notice to the Contractor within 30 Days provided that the Government gives the Contractor a preliminary written notice of its intent to extend at least 30 days before the contract expires. The preliminary notice does not commit the Government to an extension. (b) If the Government exercises this option, the extended contract shall be considered to include this option clause. (c) The total duration of this contract, including the exercise of any options under this clause, shall not exceed 8 years. ARTICLE I.3. ADDITIONAL HHSAR CLAUSES – IN FULL TEXT 352.231-70 Salary rate limitation (December 2015) (a) The Contractor shall not use contract funds to pay the direct salary of an individual at a rate in excess of the Federal Executive Schedule Level II in effect on the date the funding was obligated. (b) For purposes of the salary rate limitation, the terms “direct salary,” “salary,” and “institutional base salary,” have the same meaning and are collectively referred to as “direct salary,” in this clause. An individual's direct salary is the annual compensation that the Contractor pays for an individual's direct effort (costs) under the contract. Direct salary excludes any income that

45 an individual may be permitted to earn outside of duties to the Contractor. Direct salary also excludes fringe benefits, overhead, and general and administrative expenses (also referred to as indirect costs or facilities and administrative costs). The salary rate limitation does not restrict the salary that an organization may pay an individual working under a Department of Health and Human Services contract or order; it merely limits the portion of that salary that may be paid with contract funds. (c) The salary rate limitation also applies to individuals under subcontracts. (d) If this is a multiple-year contract or order, it may be subject to unilateral modification by the Contracting Officer to ensure that an individual is not paid at a rate that exceeds the salary rate limitation provision established in the HHS appropriations act used to fund this contract. (e) See the salaries and wages pay tables on the Office of Personnel Management website for Federal Executive Schedule salary levels.

46 PART III - LIST OF DOCUMENTS, EXHIBITS AND OTHER ATTACHMENTS SECTION J - LIST OF ATTACHMENTS The following documents are attached and incorporated in this contract: 1. Statement of Work, dated September 30, 2016, 10 pages 2. Invoice/Financing Instructions for Cost-Reimbursement Type Contracts 3. Invoice Instructions for Fixed-Priced Type Contracts 4. Sample Invoice Form 5. Research Patient Care Costs 6. Report of Government Owned, Contractor Held Property, 1 page. 7. Form SF-LLL, Disclosure of Lobbying Activities, 2 pages 8. Inclusion Enrollment Report, 5/01 (Modified OAMP: 10/01), 1 page

September 29, 2016 Page 1 ATTACHMENT 1: STATEMENT OF WORK NEXT GENERATION ANTHRAX VACCINE RFP 16-100-SOL-0015 AV7909 Anthrax Vaccine 1.1 Contractual Statement of Work 1.2Scope The scope of work for this contract includes AV7909 development activities through licensure that fall into the following areas: program management, nonclinical, clinical, regulatory, and chemistry, manufacturing, and controls (CMC). The scope of work also includes activities to support post-marketing requirements. 1.3 Objective The objective of this Statement of Work (SOW) is to conduct all necessary activities to advance the development of AV7909 through Biologics License Application (BLA) submission and approval and post- marketing requirements. Activities to meet the objective of this SOW fall in three separate contract line item number (CLIN): • CLIN 0001 - Approval of Emergency Use Authorization (EUA), licensure, approval, and clearance of product through the FDA (Base) • CLIN 0001A – Conduct of a Phase 2 clinical [**] study or other studies required by the FDA [**] (Option) • CLIN 0002 - Initial purchase, storage, and delivery of product (Base) • CLIN 0003 – Phase 4 post marketing requirements (Option) • CLIN 0004 - Surge Capacity – Additional procurement of product (Option) 1.3 CLIN 0001 - Approval of Emergency Use Authorization (EUA), licensure, approval, and clearance of product through the FDA (Base) This section identifies representative tasks and sub-tasks for CLIN 0001 with associated WBS code for each task or subtask. [**] Program Management Emergent shall provide program management activities. The activities shall include but are not limited to: • Identification of and management to, distinct stages of the product development pathway that are gates for Go/No Go decisions for advancing to the next stage of the Integrated Product Development Plan. • Establishment of and tracking of milestones and timelines for the initiation conduct, and completion of product development activities for each stage with a budget (in direct costs) linked to each stage. • Ongoing evaluation of qualitative and quantitative criteria and accompanying data used to assess the scientific merit and technical feasibility of proceeding to the next stage of product development. Preamble to the Statement of Work Independently and not as an agent of the Government, the Contractor shall be required to furnish all the necessary services, qualified personnel, material, equipment, and facilities, not otherwise provided by the Government, as needed to perform the Statement of Work submitted in response to RFP 16-100-SOL- 00015.

September 29, 2016 Page 2 • Maintaining and managing staff (in-house and contracted) to assure the necessary expertise and dedicated effort to perform the work. • Directing and overseeing subcontractors and consultants to assure successful performance of planned activities within the cost and schedule constraints of the contract. • Conducting performance measurement that shall include establishing an initial plan; defining measurable parameters; defining how these parameters relate to cost and schedule impacts; their approach in providing a detailed schedule that generates a critical path for the project; and a description of the cost-accounting system used or intended to be used based on budget estimates to monitor all costs related to the contract award for both Emergent and subcontractors on a real time basis. • Manage contract activities in accordance with Earned Value Management. In this regard, Emergent shall: o Provide an Integrated Master Project Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to support product approval. The Integrated Master Project Plan shall outline key, critical path milestones, with “Go/No-Go” decision criteria and a contract Work Breakdown Structure (due within 90 days of contract award with updates as requested by the Contracting Officer’s Representative (COR). o Submit an updated Integrated Master Schedule in an approved format. o Use principles of Earned Value Management System (EVMS) in the management of this contract. o Submit a plan for a Performance Measurement Baseline Review (PMBR) electronically via email to the Contracting Officer (CO) and COR for a PBMR to occur within 90 days of contract award. • Develop and maintain a risk management plan. • Participate in regular meetings to coordinate and oversee the contracting effort. [**] Non-Clinical Toxicology Emergent shall conduct safety and toxicology of AV7909 using animal models following Good Laboratory Practice guidelines (GLP: as defined in the U.S. Code of Federal Regulations, 21CFR Part 58), as appropriate. The activities shall include but are not limited to: [**] [**] Non-Clinical Efficacy Emergent shall conduct efficacy, pharmacokinetics/pharmacodynamics, bioavailability, solubility, formulation, dose, route and schedule of the medical countermeasure using both in vitro and animal models following Good Laboratory Practice guidelines (GLP: as defined in the U.S. Code of Federal Regulations, 21 CFR Part 58), as appropriate. The activities shall include but are not limited to: [**] [**] Clinical Evaluation Emergent shall design and conduct Phase 2 and Phase 3 clinical studies in accordance with all Federal regulations and Good Clinical Practice (GCP) guidelines. The activities shall include but are not limited to:

September 29, 2016 Page 3 [**] [**] Regulatory Activities Emergent shall conduct all required regulatory activities to support submission of BLA licensure for AV7909. The activities shall include but are not limited to: [**] [**] - Chemistry and Manufacturing Controls (CMC) Emergent shall complete the manufacturing activities necessary to support BLA submission. The activities shall include but are not limited to: [**] 1.4CLIN 0001A - Conduct of a Phase 2 clinical [**] study or other studies required by the FDA [**] (Option) This section identifies representative tasks and sub-tasks for CLIN 0001A with associated WBS code for each task or subtask. [**] Program Management Emergent shall provide program management activities. The activities shall include but are not limited to: • Identification of and management to, distinct stages of the product development pathway that are gates for Go/No Go decisions for advancing to the next stage of the Integrated Product Development Plan. • Establishment of and tracking of milestones and timelines for the initiation conduct, and completion of product development activities for each stage with a budget (in direct costs) linked to each stage. • Ongoing evaluation of qualitative and quantitative criteria and accompanying data used to assess the scientific merit and technical feasibility of proceeding to the next stage of product development.

September 29, 2016 Page 4 • Maintaining and managing staff (in-house and contracted) to assure the necessary expertise and dedicated effort to perform the work. • Directing and overseeing subcontractors and consultants to assure successful performance of planned activities within the cost and schedule constraints of the contract. • Conducting performance measurement that shall include establishing an initial plan; defining measurable parameters; defining how these parameters relate to cost and schedule impacts; their approach in providing a detailed schedule that generates a critical path for the project; and a description of the cost-accounting system used or intended to be used based on budget estimates to monitor all costs related to the contract award for both Emergent and subcontractors on a real time basis. • Manage contract activities in accordance with Earned Value Management. In this regard, Emergent shall: o Provide an Integrated Master Project Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to support product approval. The Integrated Master Project Plan shall outline key, critical path milestones, with “Go/ No Go” decision criteria and a contract Work Breakdown Structure (due within 90 days of contract award with updates as requested by the Contracting Officer’s Representative (COR). o Submit an updated Integrated Master Schedule in an approved format. o Use principles of Earned Value Management System (EVMS) in the management of this contract. o Submit a plan for a Performance Measurement Baseline Review (PMBR) electronically via email to the Contracting Officer (CO) and COR for a PBMR to occur within 90 days of contract award. • Develop and maintain a risk management plan. • Participate in regular meetings to coordinate and oversee the contracting effort. [**] Clinical Evaluation Emergent shall design and conduct a Phase 2 clinical study in accordance with all Federal regulations and Good Clinical Practice (GCP) guidelines unless other studies are required by the FDA [**]. The activities shall include, but are not limited to: • [**] - AVA.214 Phase 2 [**] Study [**] - Chemistry and Manufacturing Controls (CMC) Emergent shall complete the manufacturing activities necessary to support AVA.214 Phase 2 [**] Study. The activities below are specific to conducting a Phase 2 [**] clinical study. If the FDA requires an alternate strategy for [**], the activities below may no longer be applicable. Upon new guidance from the FDA, Emergent will update the SOW accordingly. [**] 1.5CLIN 0002 - Initial purchase, storage, and delivery of product (Base) Emergent shall deliver 2,000,000 doses of AV7909 within [**] after EUA pre-authorization approval by FDA.

September 29, 2016 Page 5 1.6 CLIN 0003 - Phase 4 post marketing requirements (Option) [**]. Program Management Emergent shall provide program management activities. The activities shall include but are not limited to: • Identification of and management to, distinct stages of the product development pathway that are gates for Go/No Go decisions for advancing to the next stage of the Integrated Product Development Plan. • Establishment of and tracking of milestones and timelines for the initiation conduct, and completion of product development activities for each stage with a budget (in direct costs) linked to each stage. • Ongoing evaluation of qualitative and quantitative criteria and accompanying data used to assess the scientific merit and technical feasibility of proceeding to the next stage of product development. • Maintaining and managing staff (in-house and contracted) to assure the necessary expertise and dedicated effort to perform the work. • Directing and overseeing subcontractors and consultants to assure successful performance of planned activities within the cost and schedule constraints of the contract. • Conducting performance measurement that shall include establishing an initial plan; defining measurable parameters; defining how these parameters relate to cost and schedule impacts; their approach in providing a detailed schedule that generates a critical path for the project; and a description of the cost-accounting system used or intended to be used based on budget estimates to monitor all costs related to the contract award for both Emergent and subcontractors on a real time basis. • Manage contract activities in accordance with Earned Value Management. In this regard, Emergent shall: o Provide an Integrated Master Project Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to support product approval. The Integrated Master Project Plan shall outline key, critical path milestones, with “Go/No Go” decision criteria and a contract Work Breakdown Structure (due within 90 days of contract award with updates as requested by the Contracting Officer’s Representative (COR). o Submit an updated Integrated Master Schedule in an approved format. o Use principles of Earned Value Management System (EVMS) in the management of this contract. o Submit a plan for a Performance Measurement Baseline Review (PMBR) electronically via email to the Contracting Officer (CO) and COR for a PBMR to occur within 90 days of contract award. • Develop and maintain a risk management plan. • Participate in regular meetings to coordinate and oversee the contracting effort. [**] 1.7CLIN 0004 - Surge Capacity – Additional procurement of product (Option) Emergent shall deliver up to 25 million dose regimens (equivalent to 50 million doses of AV7909). This option may be triggered after EUA pre-authorization approval by FDA, which is currently linked to release of PPQ lots, and deliveries will start within [**] after trigger. 1.8Reporting Requirements and Deliverables Reports As part of the work to be performed under this contract, Emergent will prepare and deliver the following reports throughout the period of performance. Monthly Technical Progress Reports On the fifteenth (15) day of each month for the previous calendar month, Emergent will submit to the COR and the CO a Technical Progress Report covering the previous calendar month. The first reporting period consists of the first full month of performance plus any fractional part of the initial month. Thereafter, the reporting period will consist of each calendar month. The frequency of Technical Progress Reporting will be determined by the CO and COR during negotiations of the contract. The format and type of Technical Progress Report and Executive Summary will be provided by the COR. The Technical Progress Reports will summarize progress for the reporting period, such as: management and administrative updates, technical progress,

September 29, 2016 Page 6 issues, proposed work, manufacturing and supply chain management, and a summary of invoices. A Technical Progress Report will not be required for the period when the same month Annual Progress Reports or a Final Report are due. Emergent will submit one copy of the Technical Progress Report electronically via e-mail to the CO and COR. Annual Progress Reports On the thirtieth (30th) calendar day following the last day of each reporting period, Emergent will submit to the COR and the CO an Annual Progress Report. The first reporting period consists of the first full year of performance plus any fractional part of the initial year. Thereafter, the reporting period shall consist of each calendar year. Annual Progress Reports will summarize progress for the reporting period, such as: management and administrative updates, technical progress, issues, proposed work, manufacturing and supply chain management, and a summary of invoices. An Annual Progress Report will not be required for the period when the Final Technical Progress Report is due. Draft Final Report and Final Report Emergent will submit the Draft Final Progress Report forty-five (45) calendar days prior to the expiration date of the contract and the Final Progress Report on or before the expiration date of the contract. These reports will include a summation of the work performed and results obtained for execution of various studies or technical work packages during the entire contract period of performance. This report will be in sufficient detail to describe comprehensively the results achieved. An electronic copy of the Draft Final Report and Final Report will be submitted to the COR and CO. FDA Regulatory Agency Correspondence, Meeting Summaries, and Submissions With regard to interactions with the FDA, Emergent shall: • Forward the initial draft minutes to BARDA within five business days of any formal meeting with the FDA or other regulatory agency, and forward the final minutes when available.

September 29, 2016 Page 7 • Forward the initial draft minutes to BARDA within five business days of any informal meeting with the FDA or other regulatory agency, and forward the final minutes when available and if applicable. • Forward the dates and times of any meeting with the FDA and other regulatory agencies to BARDA as soon as the meeting times are known and make arrangements for appropriate BARDA staff to attend the meetings. • Provide BARDA the opportunity to review and comment upon any documents to be submitted to the FDA or other regulatory agency. Emergent will provide BARDA with five (5) business days in which to review and provide comments prior to Emergent’s submission to the FDA. Emergent will notify the COR and CO within 24 hours of all FDA arrivals to conduct site visits/audits by any regulatory agency and provide the USG with an exact copy (non-redacted) of the FDA Form 483 and the Establishment Inspection Report (EIR). Emergent will provide the COR and CO copies of the plan for addressing areas of non-conformance to FDA regulations for Good Laboratory Practice (GLP) guidelines as identified in the audit report, status updates during the plans execution, and a copy of all final responses to the FDA. Emergent will also provide redacted copies of any FDA audits received from subcontractors that occur as a result of this contract or for this product. Emergent will make arrangements with the COR for the appropriate BARDA representative(s) to be present during the final debrief by the regulatory inspector.

September 29, 2016 Page 8 Key Deliverables A summary of Key Deliverables for this contract follow No. Deliverable Description Due Date 01 Monthly Progress Report Shall include a description of the activities during the reporting period and the activities planned for the ensuing reporting period. The first reporting period consists of the first full month of performance plus any fractional part of the initial month. Thereafter, the reporting period shall consist of each calendar month. Due on or before the 15th day of each month following the end of each reporting period. Monthly progress reports are not required in the same month Annual Progress reports or a Final Report are due. 02 Annual Progress Report Shall include a summation of the activities during the reporting period, and the activities planned for the ensuing reporting period. The first reporting period consists of the first full year of performance plus any fractional part of the initial year. Thereafter, the reporting period shall consist of each calendar year. Due on or before the 30th calendar day following the end of each reporting period. 03 Draft Final Progress Report To include a summation of the work performed and results obtained for execution of various studies or technical work packages during entire contract period of performance. Shall be in sufficient detail to describe comprehensively the results achieved. Due 45 Calendar days prior to the expiration date of the contract. 04 Final Progress Report To include a summation of the work performed and results obtained for execution of various studies or technical work packages during entire contract period of performance. Shall be in sufficient detail to describe comprehensively the results achieved. Due on/before the expiration date of the contract. 05 FDA/Regulatory Agency Correspondence and Meeting Minutes The Contractor shall forward initial draft minutes and final draft minutes of any formal or informal meeting with the FDA or other regulatory agency. The contractor shall forward the dates and times of any meeting with the FDA and other regulatory agencies as soon as the meeting times are known and make arrangements for appropriate BARDA staff to attend the meetings. The Contractor shall provide BARDA the opportunity to review and comment upon any documents to be submitted to the FDA or other regulatory agency. The Contractor shall forward SOPs upon request from the COR. The contractor shall notify the COR and CO within 24 hours of all FDA arrivals to conduct sitevisits/audits by any regulatory agency, and provide copies of any associated reports, documentation, or communication. Due within 5 business days of each meeting for Contractor’s minutes, upon receipt of minutes from FDA/ regulatory agency, and upon request from the COR or Co-COR. 06 Integrated Master Project Plan (Critical Path Milestones, Work Breakdown Structure, Risk Mitigation Plan/ Matrix) The contractor shall provide an Integrated Master Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to annual deliverables (key, critical path milestones, with “Go/No Go” decision criteria) and Work Breakdown Structure (WBS) elements that shall be discernable and consistent. The contractor shall develop and maintain a risk management plan that highlights potential problems and/or issues that may arise during the life of the contract, their impact on cost, schedule and performance, and appropriate remediation plans. Due within 90 days of contract award. Updates are due as requested by the COR or Co-COR. 07 Technology Packages Technology packages developed under the contract that includes complete protocols must be submitted at the request of the BARDA COR. Due upon request from the COR or Co-COR.

September 29, 2016 Page 9 No. Deliverable Description Due Date 08 Experimental Protocols The Contractor shall submit to the COR all study/experiment/test plans, designs, and protocols prior to execution for BARDA approval or upon request by the COR or Co-COR when required. Due upon request from the COR or Co-COR. 09 Annual/Final Invention Report All reports and documentation required by FAR Clause 52.227-11, Patent Rights- Ownership by the Contractor, including, but not limited to, the invention disclosure report, the confirmatory license, and the Government support certification. If no invention is disclosed or no activity has occurred on a previously disclosed invention during the applicable reporting period, a negative report shall be submitted to the CO. Annual Invention Report Due on or before the 30th calendar day after the completion of each reporting period. Final Invention Report due on or before the expiration of the contract. 10 Publications Any manuscript or scientific meeting abstract containing data generated under this contract must be submitted to COR for review prior to submission. Due within 30 calendar days for manuscripts prior to publication and 15 calendar days for abstracts. 11 Press Releases The Contractor agrees to accurately and factually represent the work conducted under this contract in all press releases. The Contractor shall ensure the CO has received and approved an advanced copy of any press release not less than five (5) business days prior to the issuance of any potential press release. Reports/Notices due for approval to the CO not less than five (5) business days prior to the issuance of any potential press release. 12 Security Report The contractor shall report to the government any activity or incident that is in violation of established security standards or indicates the loss or theft of government products Due within 24 hours after occurrence of an activity or incident. 13 Earned Value Management System Requirements Subject to the requirements under FAR 52.234-4 Earned Value Management System, the Contract shall use principles of Earned Value Management System (EVMS) in the management of this contract (include this plan as part of the monthly, annual, and final reports). The Contractor shall also submit a Performance Measurement Baseline Review plan electronically via email to the CO and COR for a PMBR to occur within 90 days of contract award, and an Integrated Master Schedule electronically via email as outlined in a format agreed upon by BARDA to the COR and CO. The Offeror shall deliver an Earned Value Contract Performance Report on a monthly basis. As detailed in Section F.3.2 Subpart F. .

September 29, 2016 Page 10 Milestone # WBS # Milestone Deliverables Summary (Details as specified in the Deliverables) Quantity Estimated Completion Date CLIN 0001 1 [**] [**] [**] 1 Electronic Copy to Contract Officer Representative (COR); 1 Electronic Copy to Contracting Officer (CO) [**] 2 [**] [**] [**] See Above [**] 3 [**] [**] [**] See Above [**] 4 [**] [**] [**] See Above [**] 5 [**] [**] [**] See Above [**] 6 [**] [**] [**] See Above [**] 7 [**] [**] [**] See Above [**] 8 [**] [**] [**] See Above [**] 9 [**] [**] [**] See Above [**] 10 [**] [**] [**] See Above [**] 11 [**] [**] [**] See Above [**] 12 [**] [**] [**] See Above [**] CLIN 0002 16 - Completion of delivery of 2 million doses of AV7909 Delivery of 2 million doses of AV7909 See Above [**]

HHSO100201600030C Emergent BioSolutions ATTACHMENT #2 INVOICE/FINANCING REQUEST INSTRUCTIONS - FOR COST-REIMBURSEMENT TYPE CONTRACTS Format: Payment requests shall be submitted on the Contractor’s self-generated form in the manner and format prescribed herein and as illustrated in the Sample Invoice/Financing Request. Standard Form 1034, Public Voucher for Purchases and Services Other Than Personal, may be used in lieu of the Contractor’s self-generated form provided it contains all of the information shown on the Sample Invoice/Financing Request. DO NOT include a cover letter with the payment request. Number of Copies: Payment requests shall be submitted in the quantity specified in the Invoice Submission Instructions in Section G of the Contract Schedule. Frequency: Payment requests shall not be submitted more frequently than once every two weeks in accordance with the Allowable Cost and Payment Clause incorporated into this contract. Small business concerns may submit invoices/financing requests more frequently than every two weeks when authorized by the Contracting Officer. Cost Incurrence Period: Costs incurred must be within the contract performance period or covered by pre-contract cost provisions. Billing of Costs Incurred: If billed costs include (1) costs of a prior billing period, but not previously billed, or (2) costs incurred during the contract period and claimed after the contract period has expired, the Contractor shall site the amount(s) and month(s) in which it incurred such costs. Contractor's Fiscal Year: Payment requests shall be prepared in such a manner that the Government can identify costs claimed with the Contractor's fiscal year. Currency: All BARDA contracts are expressed in United States dollars. When the Government pays in a currency other than United States dollars, billings shall be expressed, and payment by the Government shall be made, in that other currency at amounts coincident with actual costs incurred. Currency fluctuations may not be a basis of gain or loss to the Contractor. Notwithstanding the above, the total of all invoices paid under this contract may not exceed the United States dollars authorized. Costs Requiring Prior Approval: Costs requiring the Contracting Officer's approval, including those set forth in an Advance Understanding in the contract, shall be identified and reference the Contracting Officer's Authorization (COA) Number. In addition, the Contractor shall show any cost set forth in an Advance Understanding as a separate line item on the payment request. Invoice/Financing Request Identification: Each payment request shall be identified as either: (a) Interim Invoice/Contract Financing Request: These are interim payment requests submitted during the contract performance period. (b) Completion Invoice: The completion invoice shall be submitted promptly upon completion of the work, but no later than one year from the contract completion date, or within 120 days after settlement of the final indirect cost rates covering the year in which the contract is physically complete (whichever date is later). The Contractor shall submit the completion invoice when all costs have been assigned to the contract and it completes all performance provisions. (c) Final Invoice: A final invoice may be required after the amounts owed have been settled between the Government and the Contractor (e.g., resolution of all suspensions and audit exceptions).

HHSO100201600030C Emergent BioSolutions Preparation and Itemization of the Invoice/Financing Request: The Contractor shall furnish the information set forth in the instructions below. The instructions are keyed to the entries on the Sample Invoice/Financing Request. (a) Designated Billing Office Name and Address: Enter the designated billing office name and address, as identified in the Invoice Submission Instructions in Section G of the Contract Schedule. (b) Contractor's Name, Address, Point of Contact, VIN, and DUNS or DUNS+4 Number: Show the Contractor's name and address exactly as they appear in the contract, along with the name, title, phone number, and e-mail address of the person to notify in the event of an improper invoice or, in the case of payment by method other than Electronic Funds Transfer, to whom payment is to be sent. Provide the Contractor’s Vendor Identification Number (VIN), and Data Universal Numbering System (DUNS) number or DUNS+4. The DUNS number must identify the Contractor’s name and address exactly as stated on the face page of the contract. When an approved assignment has been made by the Contractor, or a different payee has been designated, provide the same information for the payee as is required for the Contractor (i.e., name, address, point of contact, VIN, and DUNS). (c) Invoice/Financing Request Number: Insert the appropriate serial number of the payment request. (d) Date Invoice/Financing Request Prepared: Insert the date the payment request is prepared. (e) Contract Number and Order Number (if applicable): Insert the contract number and order number (if applicable). (f) Effective Date: Insert the effective date of the contract or if billing under an order, the effective date of the order. (g) Total Estimated Cost of Contract/Order: Insert the total estimated cost of the contract, exclusive of fixed-fee. If billing under an order, insert the total estimated cost of the order, exclusive of fixed- fee. For incrementally funded contracts/orders, enter the amount currently obligated and available for payment. (h) Total Fixed-Fee: Insert the total fixed-fee (where applicable) or the portion of the fixed-fee applicable to a particular invoice as defined in the contract. (i) Two-Way/Three-Way Match: Identify whether payment is to be made using a two-way or three- way match. To determine required payment method, refer to the Invoice Submission Instructions in Section G of the Contract Schedule. (j) Office of Acquisitions: Insert the name of the Office of Acquisitions, as identified in the Invoice Submission Instructions in Section G of the Contract Schedule. (k) Central Point of Distribution: Insert the Central Point of Distribution, as identified in the Invoice Submission Instructions in Section G of the Contract Schedule. (l) Billing Period: Insert the beginning and ending dates (month, day, and year) of the period in which costs were incurred and for which reimbursement is claimed. (m) Amount Billed - Current Period: Insert the amount claimed for the current billing period by major cost element, including any adjustments and fixed-fee. If the Contract Schedule contains separately

HHSO100201600030C Emergent BioSolutions priced line items, identify the contract line item(s) on the payment request and include a separate breakdown (by major cost element) for each line item. (n) Amount Billed - Cumulative: Insert the cumulative amounts claimed by major cost element, including any adjustments and fixed-fee. If the Contract Schedule contains separately priced line items, identify the contract line item(s) on the payment request and include a separate breakdown (by major cost element) for each line item. (o) Direct Costs: Insert the major cost elements. For each element, consider the application of the paragraph entitled "Costs Requiring Prior Approval" on page 1 of these instructions. (1) Direct Labor: Include salaries and wages paid (or accrued) for direct performance of the contract. List individuals by name, title/position, hourly/annual rate, level of effort (actual hours or % of effort), breakdown by task performed by personnel, and amount claimed. (2) Fringe Benefits: List any fringe benefits applicable to direct labor and billed as a direct cost. Do not include in this category fringe benefits that are included in indirect costs. (3) Accountable Personal Property: Include any property having a unit acquisition cost of $5,000 or more, with a life expectancy of more than two years, and sensitive property regardless of cost (see the HHS Contractor's Guide for Control of Government Property)(e.g. personal computers). Note this is not permitted for reimbursement without pre-authorization from the CO. On a separate sheet of paper attached to the payment request, list each item for which reimbursement is requested. Include reference to the following (as applicable): - Item number for the specific piece of equipment listed in the Property Schedule, and - COA number, if the equipment is not covered by the Property Schedule. The Contracting Officer may require the Contractor to provide further itemization of property having specific limitations set forth in the contract. (4) Materials and Supplies: Include all consumable material and supplies regardless of amount. Detailed line- item breakdown (e.g. receipts, quotes, etc.) is required. (5) Premium Pay: List remuneration in excess of the basic hourly rate. (6) Consultant Fee: List fees paid to consultants. Identify consultant by name or category as set forth in the contract or COA, as well as the effort (i.e., number of hours, days, etc.) and rate billed. (7) Travel: Include domestic and foreign travel. Foreign travel is travel outside of Canada, the United States and its territories and possessions. However, for an organization located outside Canada, the United States and its territories and possessions, foreign travel means travel outside that country. Foreign travel must be billed separately from domestic travel. (8) Subcontract Costs: List subcontractor(s) by name and amount billed. Provide subcontract invoices/receipts as backup documentation. If subcontract is of the cost-reimbursement variety, detailed breakdown will be required. Regardless, include backup documentation (e.g. subcontractor invoices, quotes, etc.).

HHSO100201600030C Emergent BioSolutions (9) Other: Include all other direct costs not fitting into an aforementioned category. If over $1,000, list cost elements and dollar amounts separately. If the contract contains restrictions on any cost element, that cost element must be listed separately. (p) Cost of Money (COM): Cite the COM factor and base in effect during the time the cost was incurred and for which reimbursement is claimed, if applicable. (q) Indirect Costs: Identify the indirect cost base (IDC), indirect cost rate, and amount billed for each indirect cost category. (r) Fixed-Fee: Cite the formula or method of computation for fixed-fee, if applicable. The fixed-fee must be claimed as provided for by the contract. (s) Total Amounts Claimed: Insert the total amounts claimed for the current and cumulative periods. (t) Adjustments: Include amounts conceded by the Contractor, outstanding suspensions, and/or disapprovals subject to appeal. (u) Grand Totals (v) Certification of Salary Rate Limitation: If required by the contract (see Invoice Submission Instructions in Section G of the Contract Schedule), the Contractor shall include the following certification at the bottom of the payment request: “I hereby certify that the salaries billed in this payment request are in compliance with the Salary Rate Limitation Provisions in Section H of the contract.” **Note the Contracting Officer may require the Contractor to submit detailed support for costs claimed on payment requests. Every cost must be determined to be allocable, reasonable, and allowable per FAR Part 31.

HHSO100201600030C Emergent BioSolutions ATTACHMENT #3 INVOICE/FINANCING REQUEST INSTRUCTIONS FOR FIXED PRICE TYPE CONTRACTS General The Contractor shall submit vouchers or invoices as prescribed herein. Format Standard Form l034, Public Voucher for Purchases and Services Other Than Personal, and Standard Form l035, Public Voucher for Purchases and Services Other than Personal--Continuation Sheet, and the payee's letterhead or self-designed form should be used to submit claims for reimbursement. Number of Copies: As indicated in the contract. Frequency Invoices submitted in accordance with the Payment Clause shall be submitted monthly upon delivery of goods or services unless otherwise authorized by the Contracting Officer. Preparation and Itemization of the Invoice The invoice shall be prepared as follows: (a) Designated Billing Office and address: HHS/ASPR/BARDA 330 Independence Ave, Room G640 Washington DC 20201 ATTN: Contracting Officer (b) Invoice Number (c) Date of Invoice (d) Contract number and date (e) Payee's name and address. Show the Contractor's name (as it appears in the contract), correct address, and the title and phone number of the responsible official to whom payment is to be sent. When an approved assignment has been made by the Contractor, or a different payee has been designated, then insert the name and address of the payee instead of the Contractor. (f) Description of goods or services, quantity, unit price, (where appropriate), and total amount. (g) Charges for freight or express shipments other than F.O.B. destination. (If shipped by freight or express and charges are more than $25, attach prepaid bill.) (h) Equipment - If there is a contract clause authorizing the purchase of any item of equipment, the final invoice must contain a statement indicating that no item of equipment was purchased or include a completed form HHS-565, Report of Capitalized Nonexpendable Equipment. Currency: Where payments are made in a currency other than United States dollars, billings on the contract shall be expressed, and payment by the United States Government shall be made, in that other currency at amounts coincident with actual costs incurred. Currency fluctuations may not be a basis of gain or loss to the Contractor. Notwithstanding the above, the total of all invoices paid under this contract may not exceed the United States dollars authorized.

HHSO100201600030C Emergent BioSolutions ATTACHMENT #4 - SAMPLE INVOICE FORM Company Name This invoice represents reimbursable costs for the period from Expenditure Category Amount Billed Current Cumulative Contract Value Direct Costs: Direct Labor Fringe Benefits 0.00% Total Labor Costs: Overhead 0.00% Travel Subcontracts Consultant Fees Materials and Supplies Other Total Direct Costs G&A Rate 0.00% Subtotal: Fixed Fee 0.0 Total Amount Claimed Adjustments Grand Total $ - I certify that all payments requested are for appropriate purposes and in accordance with the contract. Name/signature of signatory authority for invoicing Designated Billing Office Name and Address: DHHS/OS/ASPR/AMCG Attn: Contracting Officer 200 C St., S.W. Washington, D.C. 20201 Contractor’s Address and Contact Information: POC: Name of accountant or COO or signatory authority for invoice Title: Phone: E-Mail: TIN: DUNS #: Invoice/Finance Number: Date Invoice Prepared: Contract No. and Title: Effective Date & Period of Performance: Total Estimated Cost of Order: Office of Acquisitions: Contracting Officer (insert name here) Office of Acquisitions Management, Contracts, Grants (AMCG) Central Point of Distribution: and

HHSO100201600030C Emergent BioSolutions ATTACHMENT #5 RESEARCH PATIENT CARE COSTS (a) Research patient care costs are the costs of routine and ancillary services provided to patients participating in research programs described in this contract. (b) Patient care costs shall be computed in a manner consistent with the principles and procedures used by the Medicare Program for determining the part of Medicare reimbursement based on reasonable costs. The Diagnostic Related Group (DRG) prospective reimbursement method used to determine the remaining portion of Medicare reimbursement shall not be used to determine patient care costs. Patient care rates or amounts shall be established by the Secretary of HHS or his duly authorized representative. (c) Prior to submitting an invoice for patient care costs under this contract, the Contractor must make every reasonable effort to obtain third party payment, where third party payors (including Government agencies) are authorized or are under a legal obligation to pay all or a portion of the charges incurred under this contract for patient care. (d) The Contractor must maintain adequate procedures to identify those research patients participating in this contract who are eligible for third party reimbursement. (e) Only those charges not recoverable from third party payors or patients and which are consistent with the terms and conditions of the contract are chargeable to this contract.

Attachment 6 REPORT OF GOVERNMENT OWNED, CONTRACTOR HELD PROPERTY CONTRACTOR: CONTRACT NUMBER: ADDRESS: REPORT DATE: ADDRESS1: ADDRESS2: FISCAL YEAR: CITY: STATE: ZIP: CLASSIFICATION BEGINNING OF PERIOD ADJUSTMENTS END OF PERIOD #ITEMS VALUE GFP ADDED CAP ADDED DELETIONS #ITEMS VALUE LAND >=$25K LAND <$25K OTHER REAL >=$25K OTHER REAL <$25K PROPERTY UNDER CONST >=$25K PROPERTY UNDER CONST <$25K PLANT EQUIP >=$25K PLANT EQUIP <$25K SPECIAL TOOLING >=$25K SPECIAL TOOLING <$25K SPECIAL TEST EQUIP >=$25K SPECIAL TEST EQUIP <$25K AGENCY PECULIAR >=$25K AGENCY PECULIAR <$25K MATERIAL >=$25K (CUMULATIVE) PROPERTY UNDER MFR >=$25K PROPERTY UNDER MFR <$25K SIGNED BY: SIGNATURE DATE SIGNED: NAME PRINTED Email TITLE TELEPHONE Report of Government Owned, Contractor Held Property (Rev 10/2014)

________ ________ _____________ _____________ _______________________ Attachment 7 DISCLOSURE OF LOBBYING ACTIVITIES Approved by OMB Complete this form to disclose lobbying activities pursuant to 31 U.S.C. 1352 0348-0046 (See reverse for public burden disclosure.) 1. Type of Federal Action: a. contract b. grant c. cooperative agreement d. loan e. loan guarantee f. loan insurance 2. Status of Federal Action: a. bid/offer/application b. initial award c. post-award 3. Report Type: a. initial filing b. material change For Material Change Only: year quarter date of last report 4. Name and Address of Reporting Entity: Prime Subawardee Tier , if known : Congressional District, if known : 5. If Reporting Entity in No. 4 is a Subawardee, Enter Name and Address of Prime: Congressional District, if known : 6. Federal Department/Agency: 7. Federal Program Name/Description: CFDA Number, if applicable :______________ 8. Federal Action Number, if known : 9. Award Amount, if known : $ 10. a. Name and Address of Lobbying Registrant b. Individuals Performing Services (including address if ( if individual, last name, first name, MI ): different from No. 10a ) (last name, first name, MI ): 11. Information requested through this form is authorized by title 31 U.S.C. section 1352. This disclosure of lobbying activities is a material representation of fact upon which reliance was placed by the tier above when this transaction was made or entered into. This disclosure is required pursuant to 31 U.S.C. 1352. This information will be available for public inspection. Any person who fails to file the required disclosure shall be subject to a civil penalty of not less than $10,000 and not more than $100,000 for each such failure. Signature: Print Name: Title: Telephone No.: Date: Federal Use Only: Authorized for Local Reproduction Standard Form LLL (Rev. 7-97) PRINT

INSTRUCTIONS FOR COMPLETION OF SF-LLL, DISCLOSURE OF LOBBYING ACTIVITIES This disclosure form shall be completed by the reporting entity, whether subawardee or prime Federal recipient, at the initiation or receipt of a covered Federal action, or a material change to a previous filing, pursuant to title 31 U.S.C. section 1352. The filing of a form is required for each payment or agreement to make payment to any lobbying entity for influencing or attempting to influence an officer or employee of any agency, a Member of Congress, an officer or employee of Congress, or an employee of a Member of Congress in connection with a covered Federal action. Complete all items that apply for both the initial filing and material change report. Refer to the implementing guidance published by the Office of Management and Budget for additional information. 1. Identify the type of covered Federal action for which lobbying activity is and/or has been secured to influence the outcome of a covered Federal action. 2. Identify the status of the covered Federal action. 3. Identify the appropriate classification of this report. If this is a follow up report caused by a material change to the information previously reported, enter the year and quarter in which the change occurred. Enter the date of the last previously submitted report by this reporting entity for this covered Federal action. 4. Enter the full name, address, city, State and zip code of the reporting entity. Include Congressional District, if known. Check the appropriate classification of the reporting entity that designates if it is, or expects to be, a prime or subaward recipient. Identify the tier of the subawardee, e.g., the first subawardee of the prime is the 1st tier. Subawards include but are not limited to subcontracts, subgrants and contract awards under grants. 5. If the organization filing the report in item 4 checks "Subawardee," then enter the full name, address, city, State and zip code of the prime Federal recipient. Include Congressional District, if known. 6. Enter the name of the Federal agency making the award or loan commitment. Include at least one organizational level below agency name, if known. For example, Department of Transportation, United States Coast Guard. 7. Enter the Federal program name or description for the covered Federal action (item 1). If known, enter the full Catalog of Federal Domestic Assistance (CFDA) number for grants, cooperative agreements, loans, and loan commitments. 8. Enter the most appropriate Federal identifying number available for the Federal action identified in item 1 (e.g., Request for Proposal (RFP) number; Invitation for Bid (IFB) number; grant announcement number; the contract, grant, or loan award number; the application/proposal control number assigned by the Federal agency). Include prefixes, e.g., "RFP-DE-90-001." 9. For a covered Federal action where there has been an award or loan commitment by the Federal agency, enter the Federal amount of the award/loan commitment for the prime entity identified in item 4 or 5. 10. (a) Enter the full name, address, city, State and zip code of the lobbying registrant under the Lobbying Disclosure Act of 1995 engaged by the reporting entity identified in item 4 to influence the covered Federal action. (b) Enter the full names of the individual(s) performing services, and include full address if different from 10 (a). Enter Last Name, First Name, and Middle Initial (MI). 11. The certifying official shall sign and date the form, print his/her name, title, and telephone number. According to the Paperwork Reduction Act, as amended, no persons are required to respond to a collection of information unless it displays a valid OMB Control Number. The valid OMB control number for this information collection is OMB No. 0348-0046. Public reporting burden for this collection of information is estimated to average 10 minutes per response, including time for reviewing instructions, searching existing data sources, gathering and maintaining the data needed, and completing and reviewing the collection of information. Send comments regarding the burden estimate or any other aspect of this collection of information, including suggestions for reducing this burden, to the Office of Management and Budget, Paperwork Reduction Project (0348-0046), Washington, DC 20503.

Cumulative Inclusion Enrollment Report This report format should NOT be used for collecting data from study participants. Study Title: Comments: Racial Categories Ethnic Categories Total Not Hispanic or Latino Hispanic or Latino Unknown/Not Reported Ethnicity Female Male Unknown/ Not Reported Female Male Unknown/ Not Reported Female Male Unknown/ Not Reported American Indian/ Alaska Native 0 Asian 0 Native Hawaiian or Other Pacific Islander 0 Black or African American 0 White 0 More Than One Race 0 Unknown or Not Reported 0 Total 0 0 0 0 0 0 0 0 0 0 PHS 398 / PHS 2590 (Rev. 08/12 Approved Through 8/31/2015) OMB No. 0925-0001/0002 Page Cumulative Inclusion Enrollment Report

Certain identified information has been excluded from the exhibit because it is both (i) not material and (ii) is the type of information that the registrant treats as private or confidential. Double asterisks denote omissions. AMENDMENT OF SOLICITATION/MODIFICATION OF CONTRACT 1. CONTRACT ID CODE PAGE OF PAGES 1 6 2. AMENDMENT/MODIFICATION NO. 0001 3. EFFECTIVE DATE See Block 16C 4. REQUISITION/PURCHASE NO. 5. PROJECT NO. (If applicable) 6. ISSUED BY CODE ASPR-BARDA 7. ADMINISTERED BY (If other than Item 6) CODE ASPR-BARDA ASPR-BARDA 200 Independence Ave., S.S. Room 640-G Washington, DC 20201 ASPR-BARDA 200 Independence Ave., S.S. Room 638-G Washington, DC 20201 8. NAME AND ADDRESS OF CONTRACTOR (No., Street, county, State and ZIP Code) (x) 9A. AMENDMENT OF SOLICITATION NO. EMERGENT PRODUCT DEVELOPMENT GAITHERSBURG INC. EMERGENT PRODUCT DEVELOPMENT GAITHE 300 PROFESSIONAL DR # 100 GAITHERSBURG MD 208793419 9B. DATED (SEE ITEM 11) x 10A. MODIFICATION OF CONTRACT/ORDER NO. HHSO100201600030C 10B. DATED (SEE ITEM 13) CODE 1365869 FACILITY CODE 09/30/2016 11. THIS ITEM ONLY APPLIES TO AMENDMENTS OF SOLICITATIONS □ The above numbered solicitation is amended as set forth in Item 14. The hour and date specified for receipt of Offers □ is extended, □ is not extended. Offers must acknowledge receipt of this amendment prior to the hour and date specified in the solicitation or as amended, by one of the following methods: (a) By completing Items 8 and 15, and returning ____ copies of the amendment; (b) By acknowledging receipt of this amendment on each copy of the offer submitted; or (c) By separate letter or telegram which includes a reference to the solicitation and amendment numbers. FAILURE OF YOUR ACKNOWLEDGMENT TO BE RECEIVED AT THE PLACE DESIGNATED FOR THE RECEIPT OF OFFERS PRIOR TO THE HOUR AND DATE SPECIFIED MAY RESULT IN REJECTION OF YOUR OFFER. If by virtue of this amendment you desire to change an offer already submitted, such change may be made by telegram or letter, provided each telegram or letter makes reference to the solicitation and this amendment, and is received prior to the opening hour and date specified. 12. ACCOUNTING AND APPROPRIATION DATA (If required) See Schedule 13. THIS ITEM APPLIES ONLY TO MODIFICATIONS OF CONTRACTS/ORDERS. IT MODIFIES THE CONTRACT/ORDER NO. AS DESCRIBED IN ITEM 14. CHECK ONE A. THIS CHANGE ORDER IS ISSUED PURSUANT TO: (Specify authority) THE CHANGES SET FORTH IN ITEM 14 ARE MADE IN THE CONTRACT ORDER NO. IN ITEM 10A. B. THE ABOVE NUMBERED CONTRACT/ORDER IS MODIFIED TO REFLECT THE ADMINISTRATIVE CHANGES (such as changes in paying office, appropriation date, etc) SET FORTH IN ITEM 14, PURSUANT TO THE AUTHORITY OF FAR 43.103(b). C. THIS SUPPLEMENTAL AGREEMENT IS ENTERED INTO PURSUANT TO AUTHORITY OF: X D. OTHER (Specify type of modification and authority) FAR 52.243-2 Changes – Cost Reimbursement E. IMPORTANT: Contractor □ is not, ⊠ is required to sign this document and return 2 copies to the issuing office. 14. DESCRIPTION OF AMENDMENT/MODIFICATION (Organized by UCF section headings, including solicitation/contract subject matter where feasible.) Tax ID Number: [**] DUNS Number: [**] The purpose of this modification is to modify ARTICLES B.2 BASE PERIOD, B.3. OPTION PRICES, B.5. ADVANCE UNDERSTANDINGS, and SECTION 1 - CONTRACT CLAUSES. Funds Obligated Prior to this Modification: $198,705,042 Funds Obligated with Mod #1: $0 Total Funds Obligated to Date: $198,705,042 Expiration Date: September 29,2021 Period of Performance: 09/30/2016 to 09/29/2021 Except as provided herein, all terms and conditions of the document referenced in Item 9A or 10A, as heretofore changed, remains unchanged and in full force and effect. 15A. NAME AND TITLE OF SIGNER (Type or print) Barbara Solow SVP Research and Development 16A. NAME OF CONTRACTING OFFICER CHRISTOPHER SCOTT 15B. CONTRACTOR/OFFEROR /s/ Barbara Solow (Signature of person authorized to sign) 15C. DATE SIGNED Mar 14, 2017 16B. UNITED STATES OF AMERICA By /s/ Christopher Scott (Signature of Contracting Officer) 16C. DATE SIGNED 3/16/17 NSN 7540-01-152-8070 STANDARD FORM 30 (Rev. 10-83) Previous edition unusable Prescribed by GSA FAR (48 CFR) 53.243

2 ARTICLE B.2. BASE PERIOD is hereby modified as follows: CLIN Period of Performance Supplies/ Services Total Est. Cost Fixed Fee (7%) Total Cost Plus Fixed Fee COST REIMBURSEMENT 0001 (Base) 09/30/2016 – 09/29/2021 Licensure, approval, and clearance of product through the FDA [**] [**] [**] FIRM FIXED PRICE CLIN Period of Performance Supplies/ Services Units (# of Doses) Unit Price ($) Total ($) 0002 (Base) 09/30/2016 – 09/29/2021 Initial Purchase, Storage, and Delivery of Product 3,000,000 [**] [**] Total CLINS 1&2 09/30/2016 – 09/29/2021 See Above Descriptions $198,705,042 (Funded) [**] ARTICLE B.3. OPTION PRICES is hereby modified as follows: CLIN Period of Performance Supplies/ Services Total Est. Cost Fixed Fee Total Cost Plus Fixed Fee ($) COST REIMBURSEMENT 0001A (Option Quantity) [**] Phase II [**] Study or studies required by the FDA [**] [**] [**] [**] CLIN Period of Performance Supplies/ Services Total Est. Cost Fixed Fee Total Cost Plus Fixed Fee ($) FIXED PRICE 0003 (Option Quantity) [**] Phase IV post marketing commitments /Requirements (This is an option that may or may not be exercised during the base period as determined by the need and as established by the FDA) N/A N/A [**] CLIN Period of Performance Supplies/ Services Units (# of Product) FY 2018 Unit Price ($) Total ($) 0004A (Option Quantity) [**] Additional Surge Capacity (EUA) 7,500,000 to [**] [**] [**]

3 0004B (Option Quantity) [**] Additional Surge Capacity (Licensure) 7,500,000 to [**] [**] [**] 0004C (Option Quantity) [**] Additional Surge Capacity (EUA) [**] [**] [**] 0004D (Option Quantity) [**] Additional Surge Capacity (Licensure) [**] [**] [**] 0004E (Option Quantity) [**] Additional Surge Capacity (EUA) [**] [**] [**] 0004F (Option Quantity) [**] Additional Surge Capacity (Licensure) [**] [**] [**] 0004G (Option Quantity) [**] Additional Surge Capacity (EUA) [**] [**] [**] 0004H (Option Quantity) [**] Additional Surge Capacity (Licensure) [**] [**] [**] [**] ARTICLE B.5. ADVANCE UNDERSTANDINGS is hereby modified as follows: h. Option CLINS If procurement for CLIN 4 occurs after FY 2018, the following chart illustrates the dose prices to be used: Units (# of Doses) FY 2019 Unit Price ($) FY 2020 Unit Price ($) FY 2021 Unit Price ($) 7,500,000 to [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**]

4 The USG reserves the right to re-negotiate the option CLINS based on availability of funds and feedback received from the FDA. SECTION I - CONTRACT CLAUSES ARTICLE I.1. FAR 52.252-2, CLAUSES INCORPORATED BY REFERENCE (FEBRUARY 1998) is hereby modified to add FAR 52.219-9 as follows: This contract incorporates the following clauses by reference, with the same force and effect as if they were given in full text. Upon request, the Contracting Officer will make their full text available. Also, the full text of a clause may be accessed electronically at these addresses: https://www.acquisition.gov/FAR/ . HHSAR Clauses at: http://www.hhs.gov/policies/hhsar/subpart352.html . General Clauses for Cost-Reimbursement/Fixed Price Research and Development Contract (1) FEDERAL ACQUISITION REGULATION (FAR) (48 CFR CHAPTER 1) CLAUSES: Reg Clause Date Clause Title FAR 52.202-1 Nov 2013 Definitions FAR 52.203-3 Apr 1984 Gratuities FAR 52.203-5 May 2014 Covenant Against Contingent Fees FAR 52.203-6 Sep 2006 Restrictions on Subcontractor Sales to the Government FAR 52.203-7 May 2014 Anti-Kickback Procedures FAR 52.203-8 May 2014 Cancellation, Rescission, and Recovery of Funds for Illegal or Improper Activity FAR 52.203-10 May 2014 Price or Fee Adjustment for Illegal or Improper Activity FAR 52.203-12 Oct 2010 Limitation on Payments to Influence Certain Federal Transactions FAR 52.203-13 Oct 2015 Contractor Code of Business Ethics and Conduct FAR 52.203-14 Oct 2015 Display of Hotline Poster(s) FAR 52.203-17 Apr 2014 Contractor Employee Whistleblower Rights and Requirement To Inform Employees of Whistleblower Rights FAR 52.204-4 May 2011 Printed or Copied Double-Sided on Postconsumer Fiber Content Paper FAR 52.204-7 Jul 2013 System for Award Management FAR 52.204-10 Oct 2015 Reporting Executive Compensation and First-Tier Subcontract Awards FAR 52.204-13 Jul 2013 System for Award Management Maintenance FAR 52.209-6 Oct 2015 Protecting the Government's Interests When Subcontracting With Contractors Debarred, Suspended, or Proposed for Debarment FAR 52.209-10 Nov 2015 Prohibition on Contracting with Inverted Domestic Corporations FAR 52.210-1 Apr 2011 Market Research FAR 52.215-2 Oct 2010 Audit and Records – Negotiation FAR 52.215-8 Oct 1997 Order of Precedence - Uniform Contract Format FAR 52.215-10 Aug 2011 Price Reduction for Defective Cost or Pricing Data FAR 52.215-11 Aug 2011 Price Reduction for Defective Certified Cost or Pricing Data—Modifications. FAR 52.215-12 Oct 2010 Subcontractor Certified Cost or Pricing Data FAR 52.215-13 Oct 2010 Subcontractor Certified Cost or Pricing Data—Modifications FAR 52.215-15 Oct 2010 Pension Adjustments and Asset Reversions FAR 52.215-17 Oct 1997 Waiver of Facilities Capital Cost of Money FAR 52.215-18 Jul 2005 Reversion or Adjustment of Plans for Postretirement Benefits (PRB) other than Pensions FAR 52.215-19 Oct 1997 Notification of Ownership Changes FAR 52.215-21 Oct 2010 Requirements for Certified Cost or Pricing Data and Data Other Than Certified Cost or Pricing Data -Modifications FAR 52.215-23 Oct 2009 Limitations on Pass-Through Charges FAR 52.216-7 Jun 2013 Allowable Cost and Payment FAR 52.216-8 Jun 2011 Fixed Fee FAR 52.219-8 Oct 2014 Utilization of Small Business Concerns

5 FAR 52.219-9 Nov 2016 Small Business Subcontracting Plan FAR 52.219-28 July 2013 Post-Award Small Business Program Representation FAR 52.222-1 Feb 1997 Notice to the Government of Labor Disputes FAR 52.222-2 Jul 1990 Payment for Overtime Premiums FAR 52.222-3 Jun2003 Convict Labor FAR 52.222-21 Apr 2015 Prohibition of Segregated Facilities FAR 52.222-26 Apr 2015 Equal Opportunity FAR 52.222-35 Oct 2015 Equal Opportunity for Veterans FAR 52.222-36 Jul 2014 Equal Opportunity for Workers with Disabilities FAR 52.222-37 Feb 2016 Employment Reports on Veterans FAR 52.222-40 Dec 2010 Notification of Employee Rights Under the National Labor Relations Act FAR 52.222-43 May 2014 Fair Labor Standards Act and Service Contract Labor Standards—Price Adjustment (Multiple Year and Option Contracts) FAR 52.222-50 Mar 2015 Combating Trafficking in Persons FAR 52.222-54 Oct 2015 Employment Eligibility Verification FAR 52.223-6 May 2001 Drug-Free Workplace FAR 52.223-18 Aug 2011 Encouraging Contractor Policy to Ban Text Messaging While Driving FAR 52.224-1 April 1984 Privacy Act Notification FAR 52.224-2 April 1984 Privacy Act FAR 52.225-13 Jun 2008 Restrictions on Certain Foreign Purchases FAR 52.227-1 Dec 2007 Authorization and Consent, Alternate 1 (APR 1984) FAR 52.227-2 Dec 2007 Notice and Assistance Regarding Patent and Copyright Infringement FAR 52.227-3 Apr 1984 Patent Indemnity FAR 52.227-11 May 2014 Patent Rights – Ownership by the Contractor FAR 52.227-14 May 2014 Rights in Data - General FAR 52.227-16 Jun 1987 Additional Data Requirements FAR 52.228-7 Mar 1996 Insurance – Liability to Third Persons FAR 52.229-3 Feb 2013 Federal, State and Local Taxes FAR 52.230-2 Oct 2015 Cost Accounting Standards FAR 52.230-6 June 2010 Administration of Cost Accounting Standards FAR 52.232-1 Apr 1984 Payments FAR 52.232-2 Apr 1984 Payments under Fixed-Price Research and Development Contracts FAR 52.232-8 Feb 2002 Discounts for Prompt Payment FAR 52.232-9 Apr 1984 Limitation on Withholding of Payments FAR 52.232-11 Apr 1984 Extras FAR 52.232-17 May 2014 Interest FAR 52.232-20 Apr 1984 Limitation of Cost FAR 52.232-23 May 2014 Assignment of Claims FAR 52.232-25 Jul 2013 Prompt Payment FAR 52.232-33 Jul 2013 Payment by Electronic Funds Transfer--System for Award Management FAR 52.233-1 May 2014 Disputes FAR 52.233-3 Aug 1996 Protest After Award, Alternate I FAR 52.233-4 Oct 2004 Applicable Law for Breach of Contract Claim FAR 52.242-1 Apr 1984 Notice of Intent to Disallow Costs FAR 52.242-3 May 2014 Penalties for Unallowable Costs FAR 52.242-4 Jan 1997 Certification of Final Indirect Costs FAR 52.242-13 Jul 1995 Bankruptcy FAR 52.243-1 Aug 1987 Changes - Fixed-Price Alternate V (Apr 1984). FAR 52.243-2 Aug 1987 Changes—Cost-Reimbursement Alternate V (Apr 1984). FAR 52.243.6 Apr 1984 Change Order Accounting FAR 52.243-7 Apr 1984 Notification of Changes FAR 52.244-2 Oct 2010 Subcontracts, Alternate 1 (Jun 2007) FAR 52.244-5 Dec 1996 Competition in Subcontracting FAR 52.244-6 Apr 2015 Subcontracts for Commercial Items FAR 52.245-1 Apr 2012 Government Property FAR 52.245-9 Apr 2012 Use and Charges

6 FAR 52.246-7 Apr 1996 Inspection of Research and Development – Fixed-Price FAR 52.246-8 May 2001 Inspection of Research and Development – Cost-Reimbursement FAR 52.246-23 Feb 1997 Limitation of Liability. FAR 52.246-25 Feb 1997 Limitation of Liability—Services FAR 52.248-1 Oct 2010 Value Engineering FAR 52.249-2 Apr 2012 Termination for the Convenience of the Government (Fixed-Price) FAR 52.249-6 May 2004 Termination (Cost-Reimbursement) FAR 52.249-8 Apr 1984 Default (Fixed-Price Supply and Service) FAR 52.249-9 Apr 1984 Default (Fixed-Price Research and Development) FAR 52.249-14 Apr 1984 Excusable Delays FAR 52.253-1 Jan 1991 Computer Generated Forms All other terms and conditions of this contract remain unchanged. End of Modification #1

Certain identified information has been excluded from the exhibit because it is both (i) not material and (ii) is the type of information that the registrant treats as private or confidential. Triple asterisks denote omissions. AMENDMENT OF SOLICITATION/MODIFICATION OF CONTRACT 1. CONTRACT ID CODE PAGE OF PAGES 1 5 2. AMENDMENT/MODIFICATION NO. 0002 3. EFFECTIVE DATE See Block 16C 4. REQUISITION/PURCHASE REQ. NO. OS226613 5. PROJECT NO. (If applicable) 6. ISSUED BY CODE ASPR-BARDA 7. ADMINISTERED BY (If other than Item 6) CODE ASPR-BARDA ASPR-BARDA 200 Independence Ave., S.W. Room 640-G Washington DC 20201 ASPR-BARDA 200 Independence Ave., S.W. Room 638-G Washington DC 20201 8. NAME AND ADDRESS OF CONTRACTOR (No., street, county, State and ZIP Code) EMERGENT PRODUCT DEVELOPMENT GAITHERSBURG INC. EMERGENT PRODUCT DEVLOPMENT GAITHE 300 PROFESSIONAL DR # 100 GAITHERSBURG MD 208793419 X X 9A. AMENDMENT OF SOLICITATION NO. 9B. DATED (See Item 11) 10A. MODIFICATION OF CONTRACT/ORDER NO. HHSO100201600030C 10B. DATED (See Item 13) CODE 1365869 FACILITY CODE 09/30/2016 11. THIS ITEM ONLY APPLIES TO AMENDMENTS OF SOLICITATIONS The above numbered solicitation is amended as set forth in Item 14. The hour and date specified for receipt of Offers is extended, is not extended. Offers must acknowledge receipt of this amendment prior to the hour and date specified in the solicitation or as amended, by one of the following methods: (a) By completing Items 8 and 15, and returning copies of the amendment; (b) By acknowledging receipt of this amendment on each copy of the offer submitted; or (c) By separate letter or telegram which includes a reference to the solicitation and amendment numbers. FAILURE OF YOUR ACKNOWLEGMENT TO BE RECEIVED AT THE PLACE DESIGNATED FOR THE RECEIPT OF OFFERS PRIOR TO THE HOUR AND DATE SPECIFIED MAY RESULT IN REJECTION OF YOUR OFFER. If by virtue of this amendment you desire to change an offer already submitted, such change may be made by telegram or letter, provided each telegram or letter makes reference to the solicitation and this amendment, and is received prior to the opening hour and date specified. 12. ACCOUNTING AND APPROPRIATION DATA (If required) Net Increase: $[***] 2018.199TWNP.25106 13. THIS ITEM APPLIES ONLY TO MODIFICATIONS OF CONTRACTS/ORDERS, IT MODIFIES THE CONTRACT/ORDER NO. AS DESCRIBED IN ITEM 14. CHECK ONE A. THIS CHANGE ORDER IS ISSUED PURSUANT TO: (Specify authority) THE CHANGES SET FORTH IN ITEM 14 ARE MADE IN THE CONTRACT ORDER NO. IN ITEM 10A. B. THE ABOVE NUMBERED CONTRACT/ORDER IS MODIFIED TO REFLECT THE ADMINISTRATIVE CHANGES (such as changes in paying office, appropriation date, etc.) SET FORTH IN ITEM 14, PURSUANT TO THE AUTHORITY OF FAR 43.103(b). X C. THIS SUPPLEMENTAL AGREEMENT IS ENTERED INTO PURSUANT TO AUTHORITY OF: FAR 52.243-2 – Changes - Cost Reimbursement D. OTHER (Specify type of modification and authority) E. IMPORTANT: Contractor is not, x is required to sign this document and return 2 copies to the issuing office. 14. DESCRIPTION OF AMENDMENT/MODIFICATION (Organized by UCF section headings, including solicitation/contract subject matter where feasible.) Tax ID Number: [***] DUNS Number: [***] The purpose of this modification is to modify ARTICLES B.3. OPTION PRICES, B.5. ADVANCE UNDERSTANDINGS, C.1. STATEMENT OF WORK, G.3. KEY PERSONNEL, and SECTION J – LIST OF ATTACHMENTS. Funds Obligated Prior to this Modification: $198,705,042 Funds Obligated with Mod #2: $[***] Total Funds Obligated to Date: $[***] Continued ... Except as provided herein, all terms and conditions of the document referenced in Item 9A or 10A, as heretofore changed, remains unchanged and in full force and effect. 15A. NAME AND TITLE OF SIGNER (Type or print) Abigail Jenkins SVP BU HEAD VAI 16A. NAME OF CONTRACTING OFFICER CHRISTOPHER SCOTT

15B. CONTRACTOR/OFFEROR /s/ Abigail Jenkins (Signature of person authorized to sign) 15C. DATE SIGNED Aug 28, 2018 16B. UNITED STATES OF AMERICA BY /s/ Christopher Scott (Signature of Contracting Officer) 16C. DATE SIGNED 08/29/2018 NSN 7540-01-152-8070 STANDARD FORM 30 (REV. 10-83) Previous Edition Unusable Prescribed by GSA FAR (48 CFR) 53.243

CONTINUATION SHEET REFERENCE NO. OF DOCUMENT BEING CONTINUED HHSO100201600030C/0002 Page 2 OF 5 NAME OF OFFEROR OR CONTRACTOR EMERGENT PRODUCT DEVELOPMENT GAITHERSBURG INC. 1365869 ITEM NO. (A) SUPPLIES/SERVICES (B) QUANTITY (C) UNIT (D) UNIT PRICE (E) AMOUNT (F) EXPIRATION DATE: September 29, 2021 (Unchanged) Delivery: 08/28/2018 Delivery Location Code: HHS/OS/ASPR HHS/OS/ASPR 200 C St SW WASHINGTON DC 20201 US Appr. Yr.: 2018 CAN: 199 TWNP Object Class: 25106 FOB: Destination Period of Performance: 09/30/2016 to 09/29/2021 Add Item 5 as follows: 5 ASPR-18-04521 – Base period funds to support a Phase II Drug-Drug Interaction study Obligated Amount: $[***] [***] NSN7540-01-152-8067 OPTIONAL FORM 336 (4-86) Sponsored by GSA FAR (48 CFR) 53.110

4 ARTICLE B.3. OPTION PRICES are hereby modified as follows: CLIN Period of Performance Supplies/ Services Total Est. Cost Fixed Fee Total Cost Plus Fixed Fee ($) COST REIMBURSEMENT 0001A (Option) [***] Phase II [**] Study or studies required by the FDA [**] [***] [***] [***] 0012 08/29/18- 09/29/21 Doxycycline Arm & Redundant Contract Filler [***] [***] [***] CLIN Period of Performance Supplies/ Services Total Est. Cost Fixed Fee Total Cost Plus Fixed Fee ($) FIXED PRICE 0003 (Option) [***] Phase IV post marketing commitments /Requirements (This is an option that may or may not be exercised during the base period as determined by the need and as established by the FDA) N/A N/A [***] CLIN Period of Performance Supplies/ Services Units (# of Product) FY 2018 Unit Price ($) Total ($) 0004 (Option) [***] Additional Surge Capacity (EUA) 7,500,000 to [***] [***] [***] 0005 (Option) [***] Additional Surge Capacity (Licensure) 7,500,000 to [***] [***] [***] 0006 (Option) [***] Additional Surge Capacity (EUA) [***] [***] [***] 0007 (Option) [***] Additional Surge Capacity (Licensure) [***] [***] [***] 0008 (Option) [***] Additional Surge Capacity (EUA) [***] [***] [***] 0009 (Option) [***] Additional Surge Capacity (Licensure) [***] [***] [***] 0010 (Option) [***] Additional Surge Capacity (EUA) [***] [***] [***] 0011 (Option) [***] Additional Surge Capacity (Licensure) [***] [***] [***] [***] **CLIN 0012 is funded with this modification ARTICLE B.5 ADVANCE UNDERSTANDINGS is hereby modified as follows: h. Option CLINS If procurement for CLINs 4-11 occurs after FY 2018, the following chart illustrates the dose prices to be used:

5 Units (# of Doses) FY 2019 Unit Price ($) FY 2020 Unit Price ($) FY 2021 Unit Price ($) 7,500,000 to [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] The USG reserves the right to re-negotiate the option CLINS based on availability of funds and feedback received from the FDA. ARTICLE C.1. STATEMENT OF WORK is hereby modified as follows: Independently and not as an agent of the Government, the Contractor shall furnish all the necessary services, qualified personnel, material, equipment, and facilities not otherwise provided by the Government as needed to perform the Statement of Work dated August 29, 2018 set forth in SECTION J - List of Attachments, attached hereto and made a part of the contract.

6 ARTICLE G.3. KEY PERSONNEL is hereby modified as follows: The key personnel specified in this contract are considered to be essential to work performance. At least 30 days prior to diverting any of the specified individuals to other programs or contracts (or as soon as possible, if an individual must be replaced, for example, as a result of leaving the employ of the Contractor), the Contractor shall notify the Contracting Officer and shall submit comprehensive justification for the diversion or replacement request (including proposed substitutions for key personnel) to permit evaluation by the Government of the impact on performance under this contract. The Contractor shall not divert or otherwise replace any key personnel without the written consent of the Contracting Officer. The Government may modify the contract to add or delete key personnel at the request of the Contractor or Government. The following individuals are considered to be essential to the work being performed hereunder: Name Position [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] [***] *Bold indicated changes in this modification SECTION J - LIST OF ATTACHMENTS is hereby modified as follows: 1. Statement of Work, dated August 29, 2018, 10 Pages

- 1 - ATTACHMENT 1: STATEMENT OF WORK NEXT GENERATION ANTHRAX VACCINE RFP 16-100-SOL-0015 AV7909 Anthrax Vaccine 1.0 Contractual Statement of Work Preamble to the Statement of Work Independently and not as an agent of the Government, the Contractor shall be required to furnish all the necessary services, qualified personnel, material, equipment, and facilities, not otherwise provided by the Government, as needed to perform the Statement of Work submitted in response to RFP 16-100-SOL-00015. 1.1 Scope The scope of work for this contract includes AV7909 development activities through licensure that fall into the following areas: program management, nonclinical, clinical, regulatory, and chemistry, manufacturing, and controls (CMC). The scope of work also includes activities to support post-marketing requirements. 1.2 Objective The objective of this Statement of Work (SOW) is to conduct all necessary activities to advance the development of AV7909 through Biologics License Application (BLA) submission and approval and post-marketing requirements. Activities to meet the objective of this SOW fall in three separate contract line item number (CLIN): • CLIN 0001 - Approval of Emergency Use Authorization (EUA), licensure, approval, and clearance of product through the FDA (Base) • CLIN 0001A - Conduct of a Phase 2 clinical [***] study or other studies required by the FDA [***] (Option) • CLIN 0002 - Initial purchase, storage, and delivery of product (Base) • CLIN 0003 - Phase 4 post marketing requirements (Option) • CLIN 0004 - Surge Capacity - Additional procurement of product (Option) 1.3 CLIN 0001 - Approval of Emergency Use Authorization (EUA) licensure, approval, and clearance of product through the EDA (Base) This section identifies representative tasks and sub-tasks for CLIN 0001 with associated WBS code for each task or subtask. [***] Program Management Emergent shall provide program management activities. The activities shall include but are not limited to:

- 2 - • Identification of and management to, distinct stages of the product development pathway that are gates for Go/No Go decisions for advancing to the next stage of the Integrated Product Development Plan. • Establishment of and tracking of milestones and timelines for the initiation conduct, and completion of product development activities for each stage with a budget (in direct costs) linked to each stage. • Ongoing evaluation of qualitative and quantitative criteria and accompanying data used to assess the scientific merit and technical feasibility of proceeding to the next stage of product development. • Maintaining and managing staff (in-house and contracted) to assure the necessary expertise and dedicated effort to perform the work. • Directing and overseeing subcontractors and consultants to assure successful performance of planned activities within the cost and schedule constraints of the contract. • Conducting performance measurement that shall include establishing an initial plan; defining measurable parameters; defining how these parameters relate to cost and schedule impacts; their approach in providing a detailed schedule that generates a critical path for the project; and a description of the cost-accounting system used or intended to be used based on budget estimates to monitor all costs related to the contract award for both Emergent and subcontractors on a real time basis. • Manage contract activities in accordance with Earned Value Management. In this regard, Emergent shall: o Provide an Integrated Master Project Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to support product approval. The Integrated Master Project Plan shall outline key, critical path milestones, with “Go/No-Go” decision criteria and a contract Work Breakdown Structure (due within 90 days of contract award with updates as requested by the Contracting Officer’s Representative (COR). o Submit an updated Integrated Master Schedule in an approved format. o Use principles of Earned Value Management System (EVMS) in the management of this contract. o Submit a plan for a Performance Measurement Baseline Review (PMBR) electronically via email to the Contracting Officer (CO) and COR for a PBMR to occur within 90 days of contract award. • Develop and maintain a risk management plan. • Participate in regular meetings to coordinate and oversee the contracting effort. [***] Non-Clinical Toxicology Emergent shall conduct safety and toxicology of AV7909 using animal models following Good Laboratory Practice guidelines (GLP: as defined in the U.S. Code of Federal Regulations, 21CFR Part 58), as appropriate. The activities shall include but are not limited to: [***] [***] Non-Clinical Efficacy

- 3 - Emergent shall conduct efficacy, pharmacokinetics/pharmacodynamics, bioavailability, solubility, formulation, dose, route and schedule of the medical countermeasure using both in vitro and animal models following Good Laboratory Practice guidelines (GLP: as defined in the U.S. Code of Federal Regulations, 21 CFR Part 58), as appropriate. The activities shall include but are not limited to: [***] [***] Clinical Evaluation Emergent shall design and conduct Phase 2 and Phase 3 clinical studies in accordance with all Federal regulations and Good Clinical Practice (GCP) guidelines. The activities shall include but are not limited to: [***] [***] Regulatory Activities Emergent shall conduct all required regulatory activities to support submission of BLA licensure for AV7909. The activities shall include but are not limited to: [***] [***] - Chemistry and Manufacturing Controls (CMC) Emergent shall complete the manufacturing activities necessary to support BLA submission. The activities shall include but are not limited to: [***] 1.4 CLIN 0001A - Conduct of a Phase 2 clinical [***] study or other studies required by the FDA [***] (Option) This section identifies representative tasks and sub-tasks for CLIN 0001A with associated WBS code for each task or subtask. [***] Program Management Emergent shall provide program management activities. The activities shall include but are not limited to: • Identification of and management to, distinct stages of the product development pathway that are gates for Go/No Go decisions for advancing to the next stage of the Integrated Product Development Plan. • Establishment of and tracking of milestones and timelines for the initiation conduct, and completion of product development activities for each stage with a budget (in direct costs) linked to each stage.

- 4 - • Ongoing evaluation of qualitative and quantitative criteria and accompanying data used to assess the scientific merit and technical feasibility of proceeding to the next stage of product development. • Maintaining and managing staff (in-house and contracted) to assure the necessary expertise and dedicated effort to perform the work. • Directing and overseeing subcontractors and consultants to assure successful performance of planned activities within the cost and schedule constraints of the contract. • Conducting performance measurement that shall include establishing an initial plan; defining measurable parameters; defining how these parameters relate to cost and schedule impacts; their approach in providing a detailed schedule that generates a critical path for the project; and a description of the cost-accounting system used or intended to be used based on budget estimates to monitor all costs related to the contract award for both Emergent and subcontractors on a real time basis. • Manage contract activities in accordance with Earned Value Management. In this regard, Emergent shall: o Provide an Integrated Master Project Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to support product approval. The Integrated Master Project Plan shall outline key, critical path milestones, with “Go/ No Go” decision criteria and a contract Work Breakdown Structure (due within 90 days of contract award with updates as requested by the Contracting Officer’s Representative (COR). o Submit an updated Integrated Master Schedule in an approved format. o Use principles of Earned Value Management System (EVMS) in the management of this contract. o Submit a plan for a Performance Measurement Baseline Review (PMBR) electronically via email to the Contracting Officer (CO) and COR for a PBMR to occur within 90 days of contract award. • Develop and maintain a risk management plan. • Participate in regular meetings to coordinate and oversee the contracting effort. [***] Clinical Evaluation Emergent shall design and conduct a Phase 2 clinical study in accordance with all Federal regulations and Good Clinical Practice (GCP) guidelines unless other studies are required by the FDA [***]. The activities shall include, but are not limited to: • [***] - AVA.214 Phase 2 [***] Study [***] - Chemistry and Manufacturing Controls (CMC) Emergent shall complete the manufacturing activities necessary to support AVA.214 Phase 2 [***] Study. The activities below are specific to conducting a Phase 2 [***] clinical study. If the FDA requires an alternate strategy for [***], the activities below may no longer be applicable. Upon new guidance from the FDA, Emergent will update the SOW accordingly. [***]

- 5 - 1.5 CLIN 0002 - Initial purchase, storage, and delivery of product (Base) Emergent shall deliver 2,000,000 doses of AV7909 within [***] after EUA pre-authorization approval by FDA. 1.6 CLIN 0003 - Phase 4 post marketing requirements (Option) [***]. Program Management Emergent shall provide program management activities. The activities shall include but are not limited to: • Identification of and management to, distinct stages of the product development pathway that are gates for Go/No Go decisions for advancing to the next stage of the Integrated Product Development Plan. • Establishment of and tracking of milestones and timelines for the initiation conduct, and completion of product development activities for each stage with a budget (in direct costs) linked to each stage. • Ongoing evaluation of qualitative and quantitative criteria and accompanying data used to assess the scientific merit and technical feasibility of proceeding to the next stage of product development. • Maintaining and managing staff (in-house and contracted) to assure the necessary expertise and dedicated effort to perform the work. • Directing and overseeing subcontractors and consultants to assure successful performance of planned activities within the cost and schedule constraints of the contract. • Conducting performance measurement that shall include establishing an initial plan; defining measurable parameters; defining how these parameters relate to cost and schedule impacts; their approach in providing a detailed schedule that generates a critical path for the project; and a description of the cost-accounting system used or intended to be used based on budget estimates to monitor all costs related to the contract award for both Emergent and subcontractors on a real time basis. • Manage contract activities in accordance with Earned Value Management. In this regard, Emergent shall: o Provide an Integrated Master Project Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to support product approval. The Integrated Master Project Plan shall outline key, critical path milestones, with “Go/No Go” decision criteria and a contract Work Breakdown Structure (due within 90 days of contract award with updates as requested by the Contracting Officer’s Representative (COR). o Submit an updated Integrated Master Schedule in an approved format. o Use principles of Earned Value Management System (EVMS) in the management of this contract.

- 6 - o Submit a plan for a Performance Measurement Baseline Review (PMBR) electronically via email to the Contracting Officer (CO) and COR for a PBMR to occur within 90 days of contract award. • Develop and maintain a risk management plan. • Participate in regular meetings to coordinate and oversee the contracting effort. [***] 1.7 CLINs 0004 - 0011 Surge Capacity - Additional procurement of product (Option) Emergent shall deliver up to 25 million dose regimens (equivalent to 50 million doses of AV7909). This option may be triggered after EUA pre-authorization approval by FDA, which is currently linked to release of PPQ lots, and deliveries will start within [***] after trigger. 1.8 Reporting Requirements and Deliverables Reports As part of the work to be performed under this contract, Emergent will prepare and deliver the following reports throughout the period of performance. Monthly Technical Progress Reports On the fifteenth (15) day of each month for the previous calendar month, Emergent will submit to the COR and the CO a Technical Progress Report covering the previous calendar month. The first reporting period consists of the first full month of performance plus any fractional part of the initial month. Thereafter, the reporting period will consist of each calendar month. The frequency of Technical Progress Reporting will be determined by the CO and COR during negotiations of the contract. The format and type of Technical Progress Report and Executive Summary will be provided by the COR. The Technical Progress Reports will summarize progress for the reporting period, such as: management and administrative updates, technical progress, issues, proposed work, manufacturing and supply chain management, and a summary of invoices. A Technical Progress Report will not be required for the period when the same month Annual Progress Reports or a Final Report are due. Emergent will submit one copy of the Technical Progress Report electronically via e-mail to the CO and COR. Annual Progress Reports On the thirtieth (30th) calendar day following the last day of each reporting period, Emergent will submit to the COR and the CO an Annual Progress Report. The first reporting period consists of the first full year of performance plus any fractional part of the initial year. Thereafter, the reporting period shall consist of each calendar year. Annual Progress Reports will summarize progress for the reporting period, such as: management and administrative updates, technical progress, issues, proposed work, manufacturing and supply chain management, and a summary of invoices. An Annual Progress Report will not be required for the period when the Final Technical Progress Report is due. Draft Final Report and Final Report

- 7 - Emergent will submit the Draft Final Progress Report forty-five (45) calendar days prior to the expiration date of the contract and the Final Progress Report on or before the expiration date of the contract. These reports will include a summation of the work performed and results obtained for execution of various studies or technical work packages during the entire contract period of performance. This report will be in sufficient detail to describe comprehensively the results achieved. An electronic copy of the Draft Final Report and Final Report will be submitted to the COR and CO. FDA Regulatory Agency Correspondence, Meeting Summaries, and Submissions With regard to interactions with the FDA, Emergent shall: • Forward the initial draft minutes to BARDA within five business days of any formal meeting with the FDA or other regulatory agency, and forward the final minutes when available. • Forward the initial draft minutes to BARDA within five business days of any informal meeting with the FDA or other regulatory agency, and forward the final minutes when available and if applicable. • Forward the dates and times of any meeting with the FDA and other regulatory agencies to BARDA as soon as the meeting times are known and make arrangements for appropriate BARDA staff to attend the meetings. • Provide BARDA the opportunity to review and comment upon any documents to be submitted to the FDA or other regulatory agency. Emergent will provide BARDA with five (5) business days in which to review and provide comments prior to Emergent’s submission to the FDA. Emergent will notify the COR and CO within 24 hours of all FDA arrivals to conduct site visits/audits by any regulatory agency and provide the USG with an exact copy (non-redacted) of the FDA Form 483 and the Establishment Inspection Report (EIR). Emergent will provide the COR and CO copies of the plan for addressing areas of non-conformance to FDA regulations for Good Laboratory Practice (GLP) guidelines as identified in the audit report, status updates during the plans execution, and a copy of all final responses to the FDA. Emergent will also provide redacted copies of any FDA audits received from subcontractors that occur as a result of this contract or for this product. Emergent will make arrangements with the COR for the appropriate BARDA representative(s) to be present during the final debrief by the regulatory inspector.

- 8 - Key Deliverables A summary of Key Deliverables for this contract follow No. Deliverable Description Due Date 01 Monthly Progress Report Shall include a description of the activities during the reporting period and the activities planned for the ensuing reporting period. The first reporting period consists of the first full month of performance plus any fractional part of the initial month. Thereafter, the reporting period shall consist of each calendar month. Due on or before the 15th day of each month following the end of each reporting period. Monthly progress reports are not required in the same month Annual Progress reports or a Final Report are due. 02 Annual Progress Report Shall include a summation of the activities during the reporting period, and the activities planned for the ensuing reporting period. The first reporting period consists of the first full year of performance plus any fractional part of the initial year. Thereafter, the reporting period shall consist of each calendar year. Due on or before the 30th calendar day following the end of each reporting period. 03 Draft Final Progress Report To include a summation of the work performed and results obtained for execution of various studies or technical work packages during entire contract period of performance. Shall be in sufficient detail to describe comprehensively the results achieved. Due 45 Calendar days prior to the expiration date of the contract. 04 Final Progress Report To include a summation of the work performed and results obtained for execution of various studies or technical work packages during entire contract period of performance. Shall be in sufficient detail to describe comprehensively the results achieved. Due on/before the expiration date of the contract. 05 FDA/Regulatory Agency The Contractor shall forward initial draft minutes and final draft Due within 5 business days of each meeting for Contractor’s

- 9 - No. Deliverable Description Due Date Correspondence and Meeting Minutes minutes of any formal or informal meeting with the FDA or other regulatory agency. The contractor shall forward the dates and times of any meeting with the FDA and other regulatory agencies as soon as the meeting times are known and make arrangements for appropriate BARDA staff to attend the meetings. The Contractor shall provide BARDA the opportunity to review and comment upon any documents to be submitted to the FDA or other regulatory agency. The Contractor shall forward SOPs upon request from the COR. The contractor shall notify the COR and CO within 24 hours of all FDA arrivals to conduct site visits/audits by any regulatory agency, and provide copies of any associated reports, documentation, or communication. minutes, upon receipt of minutes from FDA/ regulatory agency, and upon request from the COR or Co-COR. 06 Integrated Master Project Plan (Critical Path Milestones, Work Breakdown Structure, Risk Mitigation Plan/ Matrix) The contractor shall provide an Integrated Master Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to annual deliverables (key, critical path milestones, with “Go/No Go” decision criteria) and Work Breakdown Structure (WBS) elements that shall be discernable and consistent. The contractor shall develop and maintain a risk management plan that highlights potential problems and/or issues that may arise during the life of the contract, their impact on cost, schedule and performance, and appropriate remediation plans. Due within 90 days of contract award. Updates are due as requested by the COR or Co-COR. 07 Technology Packages Technology packages developed under the contract that includes Due upon request from the COR or Co-COR.

- 10 - No. Deliverable Description Due Date complete protocols must be submitted at the request of the BARDA COR. 08 Experimental Protocols The Contractor shall submit to the COR all study/experiment/test plans, designs, and protocols prior to execution for BARDA approval or upon request by the COR or Co-COR when required. Due upon request from the COR or Co-COR. 09 Annual/Final Invention Report All reports and documentation required by FAR Clause 52.227-11, Patent Rights-Ownership by the Contractor, including, but not limited to, the invention disclosure report, the confirmatory license, and the Government support certification. If no invention is disclosed or no activity has occurred on a previously disclosed invention during the applicable reporting period, a negative report shall be submitted to the CO. Annual Invention Report Due on or before the 30th calendar day after the completion of each reporting period. Final Invention Report due on or before the expiration of the contract. 10 Publications Any manuscript or scientific meeting abstract containing data generated under this contract must be submitted to COR for review prior to submission. Due within 30 calendar days for manuscripts prior to publication and 15 calendar days for abstracts. 11 Press Releases The Contractor agrees to accurately and factually represent the work conducted under this contract in all press releases. The Contractor shall ensure the CO has received and approved an advanced copy of any press release not less than five (5) business days prior to the issuance of any potential press release. Reports/Notices due for approval to the CO not less than five (5) business days prior to the issuance of any potential press release. 12 Security Report The contractor shall report to the government any activity or incident that is in violation of established security standards or indicates the Due within 24 hours after occurrence of an activity or incident.

- 11 - No. Deliverable Description Due Date loss or theft of government products 13 Earned Value Management System Requirements Subject to the requirements under FAR 52.234-4 Earned Value Management System, the Contract shall use principles of Earned Value Management System (EVMS) in the management of this contract (include this plan as part of the monthly, annual, and final reports). The Contractor shall also submit a Performance Measurement Baseline Review plan electronically via email to the CO and COR for a PMBR to occur within 90 days of contract award, and an Integrated Master Schedule electronically via email as outlined in a format agreed upon by BARDA to the COR and CO. The Offeror shall deliver an Earned Value Contract Performance Report on a monthly basis. As detailed in Section F.3.2 Subpart F.

Milestone # WBS # Milestone Deliverables Summary (Details as specified in the Deliverables) Quantity Estimated Completion Date CLIN 0001 1 [***] [***] [***] 1 Electronic Copy to Contract Officer Representative (COR); 1 Electronic Copy to Contracting Officer (CO) [***] 2 [***] [***] [***] See Above [***] 3 [***] [***] [***] See Above [***] 4 [***] [***] [***] See Above [***] 5 [***] [***] [***] See Above [***] 6 [***] [***] [***] See Above [***] 7 [***] [***] [***] See Above [***] 8 [***] [***] [***] See Above [***] 9 [***] [***] [***] See Above [***] 10 [***] [***] [***] See Above [***] 11 [***] [***] [***] See Above [***] 12 [***] [***] [***] See Above [***] CLIN 0002 16 [***] [***] [***] See Above [***]

Certain identified information has been excluded from the exhibit because it is both (i) not material and (ii) is the type of information that the registrant treats as private or confidential. Double asterisks denote omissions. AMENDMENT OF SOLICITATION/MODIFICATION OF CONTRACT 1. CONTRACT ID CODE PAGE OF PAGES 1 4 2. AMENDMENT/MODIFICATION NO. P00011 3. EFFECTIVE DATE See Block 16C 4. REQUISITION/PURCHASE NO. OS285761 5. PROJECT NO. (If applicable) 6. ISSUED BY CODE 7. ADMINISTERED BY (If other than Item 6) CODE ASPR-BARDA ASPR-BARDA 200 Independence Ave., S.W. Room 640-G Washington DC 20201 ASPR-BARDA 200 Independence Ave., S.W. Room 638-G Washington DC 20201 8. NAME AND ADDRESS OF CONTRACTOR (No., Street, county, State and ZIP Code) 9A. AMENDMENT OF SOLICITATION NO. EMERGENT PRODUCT DEVELOPMENT GAITHERSBURG INC. EMERGENT PRODUCT DEVELOPMENT GAITHE 300 PROFESSIONAL DR # 100 GAITHERSBURG MD 208793419 9B. DATED (SEE ITEM 11) x 10A. MODIFICATION OF CONTRACT/ORDER NO. HHSO100201600030C 10B. DATED (SEE ITEM 13) CODE 1365869 FACILITY CODE 09/30/2016 11. THIS ITEM ONLY APPLIES TO AMENDMENTS OF SOLICITATIONS The above numbered solicitation is amended as set forth in Item 14. The hour and date specified for receipt of Offers is extended, is not extended. Offers must acknowledge receipt of this amendment prior to the hour and date specified in the solicitation or as amended, by one of the following methods: (a) By completing Items 8 and 15, and returning _________ copies of the amendment; (b) By acknowledging receipt of this amendment on each copy of the offer submitted; or (c) By separate letter or electronic communication which includes a reference to the solicitation and amendment numbers. FAILURE OF YOUR ACKNOWLEDGMENT TO BE RECEIVED AT THE PLACE DESIGNATED FOR THE RECEIPT OF OFFERS PRIOR TO THE HOUR AND DATE SPECIFIED MAY RESULT IN REJECTION OF YOUR OFFER. If by virtue of this amendment you desire to change an offer already submitted, such change may be made by letter or electronic communication, provided each letter or electronic communication makes reference to the solicitation and this amendment, and is received prior to the opening hour and date specified. 12. ACCOUNTING AND APPROPRIATION DATA (If required) Net Increase: $398,550,000.00 See Schedule 13. THIS ITEM APPLIES ONLY TO MODIFICATIONS OF CONTRACTS/ORDERS, IT MODIFIES THE CONTRACT/ORDER NO. AS DESCRIBED IN ITEM 14. CHECK ONE A. THIS CHANGE ORDER IS ISSUED PURSUANT TO: (Specify authority) THE CHANGES SET FORTH IN ITEM 14 ARE MADE IN THE CONTRACT ORDER NO. IN ITEM 10A. B. THE ABOVE NUMBERED CONTRACT/ORDER IS MODIFIED TO REFLECT THE ADMINISTRATIVE CHANGES (such as changes in paying office, appropriation date, etc) SET FORTH IN ITEM 14, PURSUANT TO THE AUTHORITY OF FAR 43.103(b). C. THIS SUPPLEMENTAL AGREEMENT IS ENTERED INTO PURSUANT TO AUTHORITY OF: X D. OTHER (Specify type of modification and authority) FAR 43.103(a) Mutual Agreement of the Parties E. IMPORTANT: Contractor is not, is required to sign this document and return ________1_____ copies to the issuing office. 14. DESCRIPTION OF AMENDMENT/MODIFICATION (Organized by UCF section headings, including solicitation/contract subject matter where feasible.) Tax ID Number: [**] DUNS Number: [**] The purpose of this modification is to modify B.3 OPTION PRICES, B.5 ADVANCE UNDERSTANDINGS, C.3 STATEMENT OF WORK, and SECTION J LIST OF ATTACHMENTS Funds Obligated Prior to this Modification: $722,692,203 Funds Obligated with Mod #11: $398,550,000 Total Funds Obligated to Date: $1,121,242,203 Expiration Date: May 31, 2025 Period of Performance: 09/30/2016 to 05/31/2025 Continued … Except as provided herein, all terms and conditions of the document referenced in Item 9 A or 10A, as heretofore changed, remains unchanged and in full force and effect. 15A. NAME AND TITLE OF SIGNER (Type or print) Adam Havey EVP, COO 16A. NAME AND TITLE OF CONTRACTING OFFICER (Type or print) JILL M. JOHNSON

15B. CONTRACTOR/OFFEROR /s/Adam Havey (Signature of person authorized to sign) 15C. DATE SIGNED Sep 30, 2021 16B. UNITED STATES OF AMERICA /s/Jill M. Johnson - S (Signature of Contracting Officer) 16C. DATE SIGNED 2021.09.30 Previous edition unusable STANDARD FORM 30 (REV. 11/2016) Prescribed by GSA FAR (48 CFR) 53.243

CONTINUATION SHEET REFERENCE NO. OF DOCUMENT BEING CONTINUED HHSO100201600030C/P00011 PAGE 2 OF 4 NAME OF OFFEROR OR CONTRACTOR EMERGENT PRODUCT DEVELOPMENT GAITHERSBURG INC. 1365869 ITEM NO. (A) SUPPLIES/SERVICES (B) QUANTITY (C) UNIT (D) UNIT PRICE (E) AMOUNT (F) 10 Add Item 10 as follows: CLIN 0010 Additional Surge Capacity (EUA) Accounting Info: 2021.1991073.26088 Appr. Yr.: 2021 CAN: 1991073 Object Class: 26088 Funded: $[**] Accounting Info: 2021.1990178.26088 Appr. Yr.: 2021 CAN: 1990178 Object Class: 26088 Funded: $[**] 398,550,000.00 OPTIONAL FORM 336 (4-86) Sponsored by GSA FAR (48 CFR) 53.110 NSN 7540-01-152-8067

HHSO100201600030C P00011 Page 4 of 4 The purpose of this modification is to modify ARTICLES B.3 OPTION PRICES, B.5 ADVANCE UNDERSTANDINGS, C.1 STATEMENT OF WORK and SECTION J – LIST OF ATTACHMENTS. ARTICLE B.3. OPTION PRICES - CLIN 0010 is modified as follows: CLIN Period of Performance Supplies/Services Doses Price per Dose Total Cost Additional Doses**** CLIN 0010 (Option Quantity)*** 09/30/2021 - 03/31/2023 Additional Surge Capacity (EUA) 4,737,000* [**] [**] Dose number TBD 10,263,000 [**] [**] TOTAL 15,000,000 [**] $398,550,000 (Funded) [**]. As of the signing of this Modification to the Contract, 20,263,000 doses have been delivered. With the exercising of CLIN 0010, 4,737,000 doses of the 15,000,000 doses will be priced at a $[**] and will fulfill Contractor’s per dose cumulative reduction obligation regarding the first 25 million doses procured under the options. All doses will be billed at the price of $[**]. **Under CLIN 0010, a total of 15,000,000 doses are expected to be procured at the unit prices stated above and may include the delivery of [**] and Additional Doses as set forth herein. CLIN 0010 pricing includes the $[**] within its agreed upon pricing for 4,737,000 doses delivered. ***CLIN 0010 is funded **** Additional Doses may be delivered to BARDA as consideration under the provision Article B.5.I. In the event Contractor delivers doses [**] (See Article B.5.I), Contractor will provide a [**]% dose-replacement equivalent of additional doses to the Government. As set forth in Article B.5.I, BARDA may accept [**] if such doses are delivered along with the appropriate number of additional doses (“Additional Doses”). Additional Doses shall be calculated as [**]% of the number of delivered [**]. ARTICLE B.5. ADVANCE UNDERSTANDINGS is hereby modified as follows: I. Stability BARDA understands that the stability testing is ongoing to support long-term stability of AV7909. The contractor will continue to perform ICH compliant stability studies on AV7909. While Contractor and BARDA believe that a [**] will be achieved, this cannot be confirmed until FDA licensure of the vaccine. For the agreed upon price, AV7909 will be delivered to the SNS that is [**] from the date of manufacture stamped on the vial label. • For CLINs 0004, 0006 and 0010, BARDA agrees to allow delivery of and may accept doses [**] if such doses are delivered along with the appropriate number of additional doses (“Additional Doses”). Additional Doses shall be calculated as [**]% of the number of delivered [**]. Contractor shall provide the Additional

HHSO100201600030C P00011 Page 5 of 4 Doses as consideration for BARDA’s acceptance of [**] with a [**] for storage in the Strategic National Stockpile. The Additional Doses will be included with delivery of the [**] at no additional cost to BARDA or the US Government. SECTION C – DESCRIPTION/SPECIFICATIONS/WORK STATEMENT ARTICLE C.1. STATEMENT OF WORK Independently and not as an agent of the Government, the Contractor shall furnish all necessary services, qualified personnel, material, equipment, and facilities not otherwise provided by the Government as needed to perform the Statement of Work dated September 30, 2021, set forth in SECTION J – List of Attachments, attached hereto and incorporated into this Prime Contract. SECTION J – LIST OF ATTACHMENTS is hereby modified as follows: 1. Statement of Work, dated September 30, 2021, 10 pages 2. AV7909 Target Delivery Schedule (CLIN 0010) Q4 2021 Q1 2022 Q2 2022 Q3 2022 Q4 2022 Q1 2023 Oct-Dec Jan-Mar Apr-Jun Jul-Sep Oct-Dec Jan-Mar [**] Doses [**] Doses [**] Doses [**] Doses [**] Doses [**] Doses All other terms and conditions of this contract remain unchanged. End of Modification #11

September 30, 2021 Page 1 ATTACHMENT 1: STATEMENT OF WORK NEXT GENERATION ANTHRAX VACCINE RFP 16-100-SOL-0015 AV7909 Anthrax Vaccine 1.0 Contractual Statement of Work Preamble to the Statement of Work Independently and not as an agent of the Government, the Contractor shall be required to furnish all the necessary services, qualified personnel, material, equipment, and facilities, not otherwise provided by the Government, as needed to perform the Statement of Work submitted in response to RFP 16-100-SOL-00015. 1.1 Scope The scope of work for this contract includes AV7909 development activities through licensure that fall into the following areas: program management, nonclinical, clinical, regulatory, and chemistry, manufacturing, and controls (CMC). The scope of work also includes activities to support post-marketing requirements. 1.2 Objective The objective of this Statement of Work (SOW) is to conduct all necessary activities to advance the development of AV7909 through Biologics License Application (BLA) submission and approval and post-marketing requirements. Activities to meet the objective of this SOW fall in seven separate contract line item number (CLIN): • CLIN 0001 – Approval of Emergency Use Authorization (EUA), licensure, approval, and clearance of product through the FDA (Base) • CLIN 0001A – Conduct of a Phase 2 clinical [**] study or other studies required by the FDA [**] (Option) • CLIN 0012 – Include doxycycline arm in the conduct of the Phase 2 clinical drug-drug interaction study and qualify a redundant contract filler (Base) • CLIN 0002 – Initial purchase, storage, and delivery of product (Base) • CLIN 0003 – Phase 4 post marketing requirements (Option) • CLIN 0004 – Surge Capacity – Additional procurement of product (EUA) (Option) – [**] to [**]1 • CLIN 0006 – Surge Capacity – Additional procurement of product (EUA) (Option) – [**] to [**] doses • CLIN 0010 – Surge Capacity - Additional procurement of product (EUA) (Option) – [**] or more doses 1.3 CLIN 0001 - Approval of Emergency Use Authorization (EUA), licensure, approval, and clearance of product through the FDA (Base) [**]

September 30, 2021 Page 2 This section identifies representative tasks and sub-tasks for CLIN 0001 with associated WBS code for each task or subtask. [**] Program Management Emergent shall provide program management activities. The activities shall include but are not limited to: • Identification of and management to, distinct stages of the product development pathway that are gates for Go/No Go decisions for advancing to the next stage of the Integrated Product Development Plan. • Establishment of and tracking of milestones and timelines for the initiation conduct, and completion of product development activities for each stage with a budget (in direct costs) linked to each stage. • Ongoing evaluation of qualitative and quantitative criteria and accompanying data used to assess the scientific merit and technical feasibility of proceeding to the next stage of product development. • Maintaining and managing staff (in-house and contracted) to assure the necessary expertise and dedicated effort to perform the work. • Directing and overseeing subcontractors and consultants to assure successful performance of planned activities within the cost and schedule constraints of the contract. • Conducting performance measurement that shall include establishing an initial plan; defining measurable parameters; defining how these parameters relate to cost and schedule impacts; their approach in providing a detailed schedule that generates a critical path for the project; and a description of the cost-accounting system used or intended to be used based on budget estimates to monitor all costs related to the contract award for both Emergent and subcontractors on a real time basis. • Manage contract activities in accordance with Earned Value Management. In this regard, Emergent shall: o Provide an Integrated Master Project Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to support product approval. The Integrated Master Project Plan shall outline key, critical path milestones, with “Go/No-Go” decision criteria and a contract Work Breakdown Structure (due within 90 days of contract award with updates as requested by the Contracting Officer’s Representative (COR). o Submit an updated Integrated Master Schedule in an approved format. o Use principles of Earned Value Management System (EVMS) in the management of this contract. o Submit a plan for a Performance Measurement Baseline Review (PMBR) electronically via email to the Contracting Officer (CO) and COR for a PBMR to occur within 90 days of contract award. • Develop and maintain a risk management plan. • Participate in regular meetings to coordinate and oversee the contracting effort. [**] Non-Clinical Toxicology

September 30, 2021 Page 3 Emergent shall conduct safety and toxicology of AV7909 using animal models following Good Laboratory Practice guidelines (GLP: as defined in the U.S. Code of Federal Regulations, 21CFR Part 58), as appropriate. The activities shall include but are not limited to: • [**] [**] Non-Clinical Efficacy Emergent shall conduct efficacy, pharmacokinetics/pharmacodynamics, bioavailability, solubility, formulation, dose, route and schedule of the medical countermeasure using both in vitro and animal models following Good Laboratory Practice guidelines (GLP: as defined in the U.S. Code of Federal Regulations, 21 CFR Part 58), as appropriate. The activities shall include but are not limited to: • [**] [**] Clinical Evaluation Emergent shall design and conduct Phase 2 and Phase 3 clinical studies in accordance with all Federal regulations and Good Clinical Practice (GCP) guidelines. The activities shall include but are not limited to: • [**] [**] Regulatory Activities Emergent shall conduct all required regulatory activities to support submission of BLA licensure for AV7909. The activities shall include but are not limited to: • [**] [**] - Chemistry and Manufacturing Controls (CMC) Emergent shall complete the manufacturing activities necessary to support BLA submission. The activities shall include but are not limited to: • [**] 1.4 CLIN 0001A - Conduct of a Phase 2 clinical [**] study or other studies required by the FDA [**] (Option) This section identifies representative tasks and sub-tasks for CLIN 0001A with associated WBS code for each task or subtask. [**] Program Management Emergent shall provide program management activities. The activities shall include but are not limited to: • Identification of and management to, distinct stages of the product development pathway that are gates for Go/No Go decisions for advancing to the next stage of the Integrated Product Development Plan.

September 30, 2021 Page 4 • Establishment of and tracking of milestones and timelines for the initiation conduct, and completion of product development activities for each stage with a budget (in direct costs) linked to each stage. • Ongoing evaluation of qualitative and quantitative criteria and accompanying data used to assess the scientific merit and technical feasibility of proceeding to the next stage of product development. • Maintaining and managing staff (in-house and contracted) to assure the necessary expertise and dedicated effort to perform the work. • Directing and overseeing subcontractors and consultants to assure successful performance of planned activities within the cost and schedule constraints of the contract. • Conducting performance measurement that shall include establishing an initial plan; defining measurable parameters; defining how these parameters relate to cost and schedule impacts; their approach in providing a detailed schedule that generates a critical path for the project; and a description of the cost-accounting system used or intended to be used based on budget estimates to monitor all costs related to the contract award for both Emergent and subcontractors on a real time basis. • Manage contract activities in accordance with Earned Value Management. In this regard, Emergent shall: o Provide an Integrated Master Project Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to support product approval. The Integrated Master Project Plan shall outline key, critical path milestones, with “Go/ No Go” decision criteria and a contract Work Breakdown Structure (due within 90 days of contract award with updates as requested by the Contracting Officer’s Representative (COR). o Submit an updated Integrated Master Schedule in an approved format. o Use principles of Earned Value Management System (EVMS) in the management of this contract. o Submit a plan for a Performance Measurement Baseline Review (PMBR) electronically via email to the Contracting Officer (CO) and COR for a PBMR to occur within 90 days of contract award. • Develop and maintain a risk management plan. • Participate in regular meetings to coordinate and oversee the contracting effort. [**] Clinical Evaluation Emergent shall design and conduct a Phase 2 clinical study in accordance with all Federal regulations and Good Clinical Practice (GCP) guidelines unless other studies are required by the FDA [**]. The activities shall include, but are not limited to: • [**] - AVA.214 Phase 2 [**] Study [**] - Chemistry and Manufacturing Controls (CMC) Emergent shall complete the manufacturing activities necessary to support AVA.214 Phase 2 [**] Study. The activities below are specific to conducting a Phase 2 [**] clinical study. If the FDA requires an alternate strategy for [**], the activities below may no longer be applicable. Upon new guidance from the FDA, Emergent will update the SOW accordingly.

September 30, 2021 Page 5 • [**] 1.5 CLIN 0012 – Include doxvcvcline arm in the conduct of the Phase 2 clinical drug-drug interaction study and qualify a redundant contract filler (Base) This section identifies representative activities of CLIN 0012 associated with CLIN0001 subtask [**] - AVA.210 Phase 2 [**] and [**] Chemistry and Manufacturing Controls: • [**] 1.6 CLIN 0002 - Initial purchase, storage, and delivery of product (Base) Under the Base Period funding Emergent shall manufacture, fill, and deliver 3,000,000 doses procured in fiscal year 2019 as an initial procurement to the Strategic National Stockpile (SNS). Emergent is approved to use management reserve funding for shipping costs associated with these deliveries. 1.7 CLIN 0003 - Phase 4 post marketing requirements (Option) [**]. Program Management Emergent shall provide program management activities. The activities shall include but are not limited to: • Identification of and management to, distinct stages of the product development pathway that are gates for Go/No Go decisions for advancing to the next stage of the Integrated Product Development Plan. • Establishment of and tracking of milestones and timelines for the initiation conduct, and completion of product development activities for each stage with a budget (in direct costs) linked to each stage. • Ongoing evaluation of qualitative and quantitative criteria and accompanying data used to assess the scientific merit and technical feasibility of proceeding to the next stage of product development. • Maintaining and managing staff (in-house and contracted) to assure the necessary expertise and dedicated effort to perform the work. • Directing and overseeing subcontractors and consultants to assure successful performance of planned activities within the cost and schedule constraints of the contract. • Conducting performance measurement that shall include establishing an initial plan; defining measurable parameters; defining how these parameters relate to cost and schedule impacts; their approach in providing a detailed schedule that generates a critical path for the project; and a description of the cost-accounting system used or intended to be used based on budget estimates to monitor all costs related to the contract award for both Emergent and subcontractors on a real time basis. • Manage contract activities in accordance with Earned Value Management. In this regard, Emergent shall: o Provide an Integrated Master Project Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to support product approval. The Integrated Master Project Plan shall outline key, critical path milestones, with “Go/No Go” decision criteria and a contract Work Breakdown Structure (due

September 30, 2021 Page 6 within 90 days of contract award with updates as requested by the Contracting Officer’s Representative (COR). o Submit an updated Integrated Master Schedule in an approved format. o Use principles of Earned Value Management System (EVMS) in the management of this contract. o Submit a plan for a Performance Measurement Baseline Review (PMBR) electronically via email to the Contracting Officer (CO) and COR for a PBMR to occur within 90 days of contract award. • Develop and maintain a risk management plan. • Participate in regular meetings to coordinate and oversee the contracting effort. [**] 1.8 CLIN 0004 through 11 - Surge Capacity – Additional procurement of product (Option) Emergent shall deliver up to 25 million dose regimens (equivalent to 50 million doses of AV7909). This option may be triggered after EUA pre-authorization approval by FDA, which is currently linked to release of PPQ lots, and deliveries will start within [**] after trigger. Under CLIN 0004, Emergent shall manufacture, fill, and deliver 10,263,000 doses procured in fiscal year 2019 as an initial procurement to the Strategic National Stockpile (SNS). [**]. Under CLIN 0006 Emergent shall manufacture, fill, and deliver 10,000,000 doses procured from August 1, 2020 through July 31, 2021, as an additional procurement to the SNS. [**]. Under CLIN 0010 Emergent shall manufacture, fill, and deliver 15,000,000 doses procured from September 30, 2021 through March 31, 2023, as an additional procurement to the SNS. [**]. For CLINs 0004, 0006 and 0010, BARDA may accept [**] if such doses are delivered along with the appropriate number of additional doses (“Additional Doses”). Additional Doses shall be calculated as [**]% of the number of delivered [**]. For delivery to the SNS, Emergent shall comply with the relevant associated activities and deliverables as outlined in the Quality Agreement (attached) as signed by Emergent, BARDA, and the SNS. Emergent shall provide appropriate documentation to BARDA for quality assurance of the final drug product delivered to the SNS and invoice appropriately. 1.9 Reporting Requirements and Deliverables Reports As part of the work to be performed under this contract, Emergent will prepare and deliver the following reports throughout the period of performance. Monthly Technical Progress Reports On the fifteenth (15) day of each month for the previous calendar month, Emergent will submit to the COR and the CO a Technical Progress Report covering the previous calendar month. The first reporting period consists of the first full month of performance plus any fractional part of the initial month. Thereafter, the reporting period will consist of each calendar month. The frequency of Technical Progress Reporting will be determined by the CO and COR during negotiations of the contract. The format and type of Technical Progress Report and Executive Summary will be provided by the COR. The Technical Progress Reports will summarize

September 30, 2021 Page 7 progress for the reporting period, such as: management and administrative updates, technical progress, issues, proposed work, manufacturing and supply chain management, and a summary of invoices. A Technical Progress Report will not be required for the period when the same month Annual Progress Reports or a Final Report are due. Emergent will submit one copy of the Technical Progress Report electronically via e-mail to the CO and COR. Annual Progress Reports On the thirtieth (30th) calendar day following the last day of each reporting period, Emergent will submit to the COR and the CO an Annual Progress Report. The first reporting period consists of the first full year of performance plus any fractional part of the initial year. Thereafter, the reporting period shall consist of each calendar year. Annual Progress Reports will summarize progress for the reporting period, such as: management and administrative updates, technical progress, issues, proposed work, manufacturing and supply chain management, and a summary of invoices. An Annual Progress Report will not be required for the period when the Final Technical Progress Report is due. Draft Final Report and Final Report Emergent will submit the Draft Final Progress Report forty-five (45) calendar days prior to the expiration date of the contract and the Final Progress Report on or before the expiration date of the contract. These reports will include a summation of the work performed and results obtained for execution of various studies or technical work packages during the entire contract period of performance. This report will be in sufficient detail to describe comprehensively the results achieved. An electronic copy of the Draft Final Report and Final Report will be submitted to the COR and CO. FDA Regulatory Agency Correspondence, Meeting Summaries, and Submissions With regard to interactions with the FDA, Emergent shall: • Forward the initial draft minutes to BARDA within five business days of any formal meeting with the FDA or other regulatory agency, and forward the final minutes when available. • Forward the initial draft minutes to BARDA within five business days of any informal meeting with the FDA or other regulatory agency, and forward the final minutes when available and if applicable. • Forward the dates and times of any meeting with the FDA and other regulatory agencies to BARDA as soon as the meeting times are known and make arrangements for appropriate BARDA staff to attend the meetings. • Provide BARDA the opportunity to review and comment upon any documents to be submitted to the FDA or other regulatory agency. Emergent will provide BARDA with five (5) business days in which to review and provide comments prior to Emergent’s submission to the FDA. Emergent will notify the COR and CO within 24 hours of all FDA arrivals to conduct site visits/audits by any regulatory agency and provide the USG with an exact copy (non-redacted) of the FDA Form 483 and the Establishment Inspection Report (EIR). Emergent will provide the COR and CO copies of the plan for addressing areas of non-conformance to FDA regulations for Good Laboratory Practice (GLP) guidelines as identified in the audit report, status updates

September 30, 2021 Page 8 during the plans execution, and a copy of all final responses to the FDA. Emergent will also provide redacted copies of any FDA audits received from subcontractors that occur as a result of this contract or for this product. Emergent will make arrangements with the COR for the appropriate BARDA representative(s) to be present during the final debrief by the regulatory inspector.

September 30, 2021 Page 9 Key Deliverables A summary of Key Deliverables for this contract follow No. Deliverable Description Due Date 01 Monthly Progress Report Shall include a description of the activities during the reporting period and the activities planned for the ensuing reporting period. The first reporting period consists of the first full month of performance plus any fractional part of the initial month. Thereafter, the reporting period shall consist of each calendar month. Due on or before the 15th day of each month following the end of each reporting period. Monthly progress reports are not required in the same month Annual Progress reports or a Final Report are due. 02 Annual Progress Report Shall include a summation of the activities during the reporting period, and the activities planned for the ensuing reporting period. The first reporting period consists of the first full year of performance plus any fractional part of the initial year. Thereafter, the reporting period shall consist of each calendar year. Due on or before the 30th calendar day following the end of each reporting period. 03 Draft Final Progress Report To include a summation of the work performed and results obtained for execution of various studies or technical work packages during entire contract period of performance. Shall be in sufficient detail to describe comprehensively the results achieved. Due 45 Calendar days prior to the expiration date of the contract. 04 Final Progress Report To include a summation of the work performed and results obtained for execution of various studies or technical work packages during entire contract period of performance. Shall be in sufficient detail to describe comprehensively the results achieved. Due on/before the expiration date of the contract. 05 FDA/Regulatory Agency Correspondence and Meeting Minutes The Contractor shall forward initial draft minutes and final draft minutes of any formal or informal meeting with the FDA or other regulatory agency. The contractor shall forward the dates and times of any meeting with the FDA and other regulatory agencies as soon as the meeting times are known and make arrangements for appropriate BARDA staff to attend the meetings. The Contractor shall provide BARDA the opportunity to review and comment upon any documents to be submitted to the FDA or other regulatory agency. Due within 5 business days of each meeting for Contractor’s minutes, upon receipt of minutes from FDA/regulatory agency, and upon request from the COR or Co-COR.

September 30, 2021 Page 10 No. Deliverable Description Due Date The Contractor shall forward SOPs upon request from the COR. The contractor shall notify the COR and CO within 24 hours of all FDA arrivals to conduct site visits/audits by any regulatory agency, and provide copies of any associated reports, documentation, or communication. 06 Integrated Master Project Plan (Critical Path Milestones, Work Breakdown Structure, Risk Mitigation Plan/Matrix) The contractor shall provide an Integrated Master Plan (including tabular and Gantt forms) to BARDA that clearly indicates the critical path to annual deliverables (key, critical path milestones, with “Go/No Go” decision criteria) and Work Breakdown Structure (WBS) elements that shall be discernable and consistent. The contractor shall develop and maintain a risk management plan that highlights potential problems and/or issues that may arise during the life of the contract, their impact on cost, schedule and performance, and appropriate remediation plans. Due within 90 days of contract award. Updates are due as requested by the COR or Co- COR. 07 Technology Packages Technology packages developed under the contract that includes complete protocols must be submitted at the request of the BARDA COR. Due upon request from the COR or Co-COR. 08 Experimental Protocols The Contractor shall submit to the COR all study/experiment/test plans, designs, and protocols prior to execution for BARDA approval or upon request by the COR or Co-COR when required. Due upon request from the COR or Co-COR. 09 Annual/Final Invention Report All reports and documentation required by FAR Clause 52.227-11, Patent Rights-Ownership by the Contractor, including, but not limited to, the invention disclosure report, the confirmatory license, and the Government support certification. If no invention is disclosed or no activity has occurred on a previously disclosed invention during the applicable reporting period, a negative report shall be submitted to the CO. Annual Invention Report Due on or before the 30th calendar day after the completion of each reporting period. Final Invention Report due on or before the expiration of the contract. 10 Publications Any manuscript or scientific meeting abstract containing data generated Due within 30 calendar days for manuscripts prior to publication

September 30, 2021 Page 11 No. Deliverable Description Due Date under this contract must be submitted to COR for review prior to submission. and 15 calendar days for abstracts. 11 Press Releases The Contractor agrees to accurately and factually represent the work conducted under this contract in all press releases. The Contractor shall ensure the CO has received and approved an advanced copy of any press release not less than five (5) business days prior to the issuance of any potential press release. Reports/Notices due for approval to the CO not less than five (5) business days prior to the issuance of any potential press release. 12 Security Report The contractor shall report to the government any activity or incident that is in violation of established security standards or indicates the loss or theft of government products Due within 24 hours after occurrence of an activity or incident. 13 Earned Value Management System Requirements Subject to the requirements under FAR 52.234-4 Earned Value Management System, the Contract shall use principles of Earned Value Management System (EVMS) in the management of this contract (include this plan as part of the monthly, annual, and final reports). The Contractor shall also submit a Performance Measurement Baseline Review plan electronically via email to the CO and COR for a PMBR to occur within 90 days of contract award, and an Integrated Master Schedule electronically via email as outlined in a format agreed upon by BARDA to the COR and CO. The Offeror shall deliver an Earned Value Contract Performance Report on a monthly basis. As detailed in Section F.3.2 Subpart F.

Milestone # WBS # Milestone Deliverables Summary (Details as specified in the Deliverables) Quantity Estimated Completion Date CLIN 0001 & CLIN 0012 1 [**] [**] [**] 1 Electronic Copy to Contract Officer Representative (COR); 1 Electronic Copy to Contracting Officer (CO) 12/19/2017 2 [**] [**] [**] See Above 1/18/2018 3 [**] [**] [**] See Above 5/24/2018 4 [**] [**] [**] See Above 11/6/2018 5 [**] [**] [**] See Above 11/8/2018 8 [**] [**] [**] See Above 3/21/2021 9 [**] [**] [**] See Above 8/18/2020 10 [**] [**] [**] See Above 12/31/2021 11 [**] [**] [**] See Above 5/12/2021 12 [**] [**] [**] See Above 12/15/2021 CLIN 0002 16 - Completion of delivery of 3 million doses of AV7909 Delivery of 3 million doses of AV7909 See Above 10/24/2019

Certain identified information has been excluded from the exhibit because it is both (i) not material and (ii) is the type of information that the registrant treats as private or confidential. Double asterisks denote omissions. AMENDMENT OF SOLICITATION/MODIFICATION OF CONTRACT 1. CONTRACT ID CODE PAGE OF PAGES 1 2 2. AMENDMENT/MODIFICATION NO. P00024 3. EFFECTIVE DATE See Block 16C 4. REQUISITION/PURCHASE NO. 5. PROJECT NO. (If applicable) 6. ISSUED BY CODE ASPR/SNS 7. ADMINISTERED BY (If other than Item 6) CODE ASPR/SNS ASPR/SNS ASPR/SNS 2945 FLOWERS ROAD ATLANTA, GA 30341 US DEPT OF HEALTH & HUMAN SERVICES ASPR/SNS 2945 FLOWERS ROAD ATLANTA, GA 30341 8. NAME AND ADDRESS OF CONTRACTOR (No., Street, county, State and ZIP Code) 9A. AMENDMENT OF SOLICITATION NO. EMERGENT BIODEFENSE OPERATIONS LANSING LLC 330303 Attn: DIANA EMERGENT BIODEFENSE OPERATIONS LANS 3500 N MARTIN LUTHER KING JR BLVD LANSING MI 489062933 9B. DATED (SEE ITEM 11) X 10A. MODIFICATION OF CONTRACT/ORDER NO. HHSD200201792634C 10B. DATED (SEE ITEM 13) CODE 330303 FACILITY CODE 12/08/2016 11. THIS ITEM ONLY APPLIES TO AMENDMENTS OF SOLICITATIONS The above numbered solicitation is amended as set forth in Item 14. The hour and date specified for receipt of Offers is extended, is not extended. Offers must acknowledge receipt of this amendment prior to the hour and date specified in the solicitation or as amended, by one of the following methods: (a) By completing Items 8 and 15, and returning _________ copies of the amendment; (b) By acknowledging receipt of this amendment on each copy of the offer submitted; or (c) By separate letter or electronic communication which includes a reference to the solicitation and amendment numbers. FAILURE OF YOUR ACKNOWLEDGMENT TO BE RECEIVED AT THE PLACE DESIGNATED FOR THE RECEIPT OF OFFERS PRIOR TO THE HOUR AND DATE SPECIFIED MAY RESULT IN REJECTION OF YOUR OFFER. If by virtue of this amendment you desire to change an offer already submitted, such change may be made by letter or electronic communication, provided each letter or electronic communication makes reference to the solicitation and this amendment, and is received prior to the opening hour and date specified. 12. ACCOUNTING AND APPROPRIATION DATA (If required) See Schedule 13. THIS ITEM APPLIES ONLY TO MODIFICATIONS OF CONTRACTS/ORDERS, IT MODIFIES THE CONTRACT/ORDER NO. AS DESCRIBED IN ITEM 14. CHECK ONE A. THIS CHANGE ORDER IS ISSUED PURSUANT TO: (Specify authority) THE CHANGES SET FORTH IN ITEM 14 ARE MADE IN THE CONTRACT ORDER NO. IN ITEM 10A. X B. THE ABOVE NUMBERED CONTRACT/ORDER IS MODIFIED TO REFLECT THE ADMINISTRATIVE CHANGES (such as changes in paying office, appropriation date, etc) SET FORTH IN ITEM 14, PURSUANT TO THE AUTHORITY OF FAR 43.103(b). C. THIS SUPPLEMENTAL AGREEMENT IS ENTERED INTO PURSUANT TO AUTHORITY OF: D. OTHER (Specify type of modification and authority) E. IMPORTANT: Contractor is not, ☒ is required to sign this document and return _________1____ copies to the issuing office. 14. DESCRIPTION OF AMENDMENT/MODIFICATION (Organized by UCF section headings, including solicitation/contract subject matter where feasible.) Tax ID Number: [**] DUNS Number: [**] This modification is issued to make the following changes to Modification P00023: 1. Delete paragraph 3 of modification P00023 in its entirety. 2. The following statement is hereby added: The total contract obligated amount to date is $[**]. See Summary of Differences below. 3. The total contract value to date is $1,153,275,629.80. 4. No further changes. Continued... Except as provided herein, all terms and conditions of the document referenced in Item 9 A or 10A, as heretofore changed, remains unchanged and in full force and effect. 15A. NAME AND TITLE OF SIGNER (Type or print) Adam Havey EVP 16A. NAME AND TITLE OF CONTRACTING OFFICER (Type or print) NATASHA Y. ROWLAND

15B. CONTRACTOR/OFFEROR (Signature of person authorized to sign) 15C. DATE SIGNED Jan26, 2021 16B. UNITED STATES OF AMERICA /s/ Natasha Rowland (Signature of Contracting Officer) 16C. DATE SIGNED 02/02/2021 Previous edition unusable STANDARD FORM 30 (REV. 11/2016) Prescribed by GSA FAR (48 CFR) 53.243

CONTINUATION SHEET REFERENCE NO. OF DOCUMENT BEING CONTINUED HHSD200201792634C/P00024 PAGE 2 OF 2 NAME OF OFFEROR OR CONTRACTOR EMERGENT PRODUCT DEVELOPMENT GAITHERSBURG INC. 1365869 SUPPLIES/SERVICES (B) Summary of Differences [**] Total contract value per ASPR-BARDA through Mod 23 [**] Less: Addition error originating in Mod 17 and reoccurring in Mod 20 [**] Less: Mod 22 incorrect beginning contract value due to $[**] adjustment [**] Less: Mod 22 order of [**] doses @ $[**] per dose [**] Less: CLIN 0001 difference in price due to [**] product being delivered [**] Less: CLIN 0002 difference in price due to [**] product being delivered [**] [**] Invoiced by Emergent through 11/30/20 [**] Remaining [**] doses from Mod 23 [**] NSN 7540-01-152-8067 OPTIONAL FORM 336 (4-86) Sponsored by GSA FAR (48 CFR) 53.110

Certain identified information has been excluded from the exhibit because it is both (i) not material and (ii) is the type of information that the registrant treats as private or confidential. Double asterisks denote omissions. AMENDMENT OF SOLICITATION/MODIFICATION OF CONTRACT 1. CONTRACT ID CODE PAGE OF PAGES 1 2 2. AMENDMENT/MODIFICATION NO. P00025 3. EFFECTIVE DATE See Block 16C 4. REQUISITION/PURCHASE NO. OS285084 5. PROJECT NO. (If applicable) 6. ISSUED BY CODE ASPR/SNS 7. ADMINISTERED BY (If other than Item 6) CODE ASPR/SNS ASPR/SNS ASPR/SNS 2945 FLOWERS ROAD ATLANTA, GA 30341 US DEPT OF HEALTH & HUMAN SERVICES ASPR/SNS 2945 FLOWERS ROAD ATLANTA, GA 30341 8. NAME AND ADDRESS OF CONTRACTOR (No., Street, county, State and ZIP Code) 9A. AMENDMENT OF SOLICITATION NO. EMERGENT BIODEFENSE OPERATIONS LANSING LLC 330303 Attn: DIANA EMERGENT BIODEFENSE OPERATIONS LANS 3500 N MARTIN LUTHER KING JR BLVD LANSING MI 489062933 9B. DATED (SEE ITEM 11) x 10A. MODIFICATION OF CONTRACT/ORDER NO. HHSD200201792634C 10B. DATED (SEE ITEM 13) CODE 330303 FACILITY CODE 12/08/2016 11. THIS ITEM ONLY APPLIES TO AMENDMENTS OF SOLICITATIONS The above numbered solicitation is amended as set forth in Item 14. The hour and date specified for receipt of Offers is extended, is not extended. Offers must acknowledge receipt of this amendment prior to the hour and date specified in the solicitation or as amended, by one of the following methods: (a) By completing Items 8 and 15, and returning _________ copies of the amendment; (b) By acknowledging receipt of this amendment on each copy of the offer submitted; or (c) By separate letter or electronic communication which includes a reference to the solicitation and amendment numbers. FAILURE OF YOUR ACKNOWLEDGMENT TO BE RECEIVED AT THE PLACE DESIGNATED FOR THE RECEIPT OF OFFERS PRIOR TO THE HOUR AND DATE SPECIFIED MAY RESULT IN REJECTION OF YOUR OFFER. If by virtue of this amendment you desire to change an offer already submitted, such change may be made by letter or electronic communication, provided each letter or electronic communication makes reference to the solicitation and this amendment, and is received prior to the opening hour and date specified. 12. ACCOUNTING AND APPROPRIATION DATA (If required) N/A 13. THIS ITEM APPLIES ONLY TO MODIFICATIONS OF CONTRACTS/ORDERS, IT MODIFIES THE CONTRACT/ORDER NO. AS DESCRIBED IN ITEM 14. CHECK ONE A. THIS CHANGE ORDER IS ISSUED PURSUANT TO: (Specify authority) THE CHANGES SET FORTH IN ITEM 14 ARE MADE IN THE CONTRACT ORDER NO. IN ITEM 10A. B. THE ABOVE NUMBERED CONTRACT/ORDER IS MODIFIED TO REFLECT THE ADMINISTRATIVE CHANGES (such as changes in paying office, appropriation date, etc) SET FORTH IN ITEM 14, PURSUANT TO THE AUTHORITY OF FAR 43.103(b). X C. THIS SUPPLEMENTAL AGREEMENT IS ENTERED INTO PURSUANT TO AUTHORITY OF: FAR 43.103 (a) (3) Bilateral D. OTHER (Specify type of modification and authority) E. IMPORTANT: Contractor is not, is required to sign this document and return __________1___ copies to the issuing office. 14. DESCRIPTION OF AMENDMENT/MODIFICATION (Organized by UCF section headings, including solicitation/contract subject matter where feasible.) Tax ID Number:[**] DUNS Number: [**] This No Cost modification is issued to: 1. Extend the ordering period for Optional CLIN 0005 - Biothrax Vaccine Doses through March 31, 2022. 2. Price per unit cost until March 31, 2022 is as follows: Dose Price - [**]: $[**]; Dose Price - [**]: $[**]. 3. No further changes. Continued ... Except as provided herein, all terms and conditions of the document referenced in Item 9 A or 10A, as heretofore changed, remains unchanged and in full force and effect. 15A. NAME AND TITLE OF SIGNER (Type or print) Paul Williams SVP, MCM-Government Business 16A. NAME AND TITLE OF CONTRACTING OFFICER (Type or print) NATASHA Y. ROWLAND

15B. CONTRACTOR/OFFEROR /s/Paul Wiliams (Signature of person authorized to sign) 15C. DATE SIGNED Sep 29, 2021 16B. UNITED STATES OF AMERICA /s/Natasha Rowland - S (Signature of Contracting Officer) 16C. DATE SIGNED 09/29/2021 Previous edition unusable STANDARD FORM 30 (REV. 11/2016) Prescribed by GSA FAR (48 CFR) 53.243

CONTINUATION SHEET REFERENCE NO. OF DOCUMENT BEING CONTINUED HHSD200201792634C/P00025 PAGE 2 OF 2 NAME OF OFFEROR OR CONTRACTOR EMERGENT BIODEFENSE OPERATIONS LANSING LLC 330303 ITEM NO. (A) SUPPLIES/SERVICES (B) QUANTITY (C) UNIT (D) UNIT PRICE (E) AMOUNT (F) 7 Period of Performance: 10/01/2021 to 03/31/2022 Add Item 7 as follows: The purpose of this Requisition is to extend the POP for CLIN 0005 COR David Kelly CAN OC Amount Obligated Amount: $0.00 1 EA 0.00 0.00 OPTIONAL FORM 336 (4-86) Sponsored by GSA FAR (48 CFR) 53.110 NSN 7540-01-152-8067

Certain identified information has been excluded from the exhibit because it is both (i) not material and (ii) is the type of information that the registrant treats as private or confidential. Double asterisks denote omissions. AMENDMENT OF SOLICITATION/MODIFICATION OF CONTRACT 1. CONTRACT ID CODE PAGE OF PAGES 1 2 2. AMENDMENT/MODIFICATION NO. P00030 3. EFFECTIVE DATE See Block 16C 4. REQUISITION/PURCHASE REQ. NO. 5. PROJECT NO. (If applicable) 6. ISSUED BY CODE ASPR-BARDA 7. ADMINISTERED BY (If other than Item 6) CODE ASPR-BARDA02 ASPR-BARDA 200 Independence Ave., S.W. Room 640-G Washington, DC 20201 ASPR-BARDA 330 Independence Ave., S.W., Rm G640 Washington, DC 20201 8. NAME AND ADDRESS OF CONTRACTOR (No., street, county, State and ZIP Code) (x) 9A. AMENDMENT OF SOLICITATION NO. EMERGENT MANUFACTURING OPERATIONS BALTIMORE LLC Attn: [**] EMERGENT MANUFACTURING OPERATIONS B 5901 E LOMBARD ST BALTIMORE MD 212246824 9B. DATED (SEE ITEM 11) x 10A. MODIFICATION OF CONTRACT/ORDER NO. HHSO100201200004I 10B. DATED (SEE ITEM 13) CODE 1410445 FACILITY CODE 06/15/2012 11. THIS ITEM ONLY APPLIES TO AMENDMENTS OF SOLICITATIONS The above numbered solicitation is amended as set forth in Item 14. The hour and date specified for receipt of Offers is extended, is not extended. Offers must acknowledge receipt of this amendment prior to the hour and date specified in the solicitation or as amended, by one of the following methods: (a) By completing Items 8 and 15, and returning _________ copies of the amendment; (b) By acknowledging receipt of this amendment on each copy of the offer submitted; or (c) By separate letter or electronic communication which includes a reference to the solicitation and amendment numbers. FAILURE OF YOUR ACKNOWLEDGMENT TO BE RECEIVED AT THE PLACE DESIGNATED FOR THE RECEIPT OF OFFERS PRIOR TO THE HOUR AND DATE SPECIFIED MAY RESULT IN REJECTION OF YOUR OFFER. If by virtue of this amendment you desire to change an offer already submitted, such change may be made by letter or electronic communication, provided each letter or electronic communication makes reference to the solicitation and this amendment, and is received prior to the opening hour and date specified. 12. ACCOUNTING AND APPROPRIATION DATA (If required) See Schedule 13. THIS ITEM APPLIES ONLY TO MODIFICATIONS OF CONTRACTS/ORDERS. IT MODIFIES THE CONTRACT/ORDER NO. AS DESCRIBED IN ITEM 14. CHECK ONE A. THIS CHANGE ORDER IS ISSUED PURSUANT TO: (Specify authority) THE CHANGES SET FORTH IN ITEM 14 ARE MADE IN THE CONTRACT ORDER NO. IN ITEM 10A. B. THE ABOVE NUMBERED CONTRACT/ORDER IS MODIFIED TO REFLECT THE ADMINISTRATIVE CHANGES (such as changes in paying office, appropriation data, etc.) SET FORTH IN ITEM 14, PURSUANT TO THE AUTHORITY OF FAR 43.103(b). C. THIS SUPPLEMENTAL AGREEMENT IS ENTERED INTO PURSUANT TO AUTHORITY OF: 43.103(a)(3) – Bilateral Modification D. OTHER (Specify type of modification and authority) E. IMPORTANT: Contractor is not, is required to sign this document and return ________ copies to the issuing office. 14. DESCRIPTION OF AMENDMENT/MODIFICATION (Organized by UCF section headings, including solicitation/contract subject matter where feasible.) Tax ID Number: [**] DUNS Number: [**] The purpose of this modification is to 1) Extend the Base Period of Performance from 9/30/21 to 9/30/22 at no additional costs to allow activity under CLIN 0003 Raxi; 2) Change the Contracting Officer; and D.3. Reporting Deliverables addressee as outlined as follows: Period of Performance: 06/15/2012 to 09/30/2022 Except as provided herein, all terms and conditions of the document referenced in Item 9A or 10A, as heretofore changed, remains unchanged and in full force and effect. 15A. NAME AND TITLE OF SIGNER (Type or print). Mark Alley Vice President 16A. NAME AND TITLE OF CONTRACTING OFFICER (Type or print) Charles Strickland 15B. CONTRACTOR/OFFEROR /s/ Mark Alley (Signature of person authorized to sign) 15C. DATE SIGNED Sep 29, 2021 16B. UNITED STATES OF AMERICA /s/ Charles P. Strickland S (Signature of Contracting Officer) 16C. DATE SIGNED STANDARD FORM 30 (Rev. 11/2016) Digitally signed by Charles P. Strickland-S Date: 2021 09 30 09 01:16’

Previous edition unusable Prescribed by GSA FAR (48 CFR) 53.243

Contract No. HHSO100201200004I Modification No. 0030 Page 3 of 3 1. This is Modification No. 0030 to Contract No. HHSO100201200004I. 2. The purpose of this modification is to: a) Extend the Base Period of Performance at no additional costs to the government to allow for activity under CLIN 0003 RAXI; 2) Change the Contracting Officer; and reporting deliverables addressee under Section D.3. b) This modification shall provide the adequate lead time to ensure the Contractor meet their obligations under CLIN 0003 RAXI at the Bayview site that was put on hold overall due to ongoing COVID activities introduced in early 2020. 3. Accordingly, the following changes are made to the contract: a) The Base Period of Performance is extended from 09/30/2021 to 09/30/2022 at no additional cost to the Government. b) Work and technical deliverables to be completed during the requested extension are outlined as follows: i. DP Tech Transfer (ongoing) ; ii. Bulk Drug Substance (BDS) Master Validation package (currently on hold); iii. Final Drug Product (FDP) Master Validation package iv. Final Master Batch Records - BDS; v. Final Master Batch Records – FDP; vi. Final validation reports (BDS and FDP); vii. Final submission and approval letter from FDA for SBLA approval; viii. Regulatory bridging plan (Comparability Plan) with Milestones; ix. Update site master file – due within [**] within PPQ completion c) No invoicing for this CLIN will occur during the extension period. 4. The Contracting Officer is changed from [**] to [**]; and accordingly [**] replaces [**] under Section D.3 Report Deliverables, with the following address: [**] Contracting Officer (CO) HHS/OS/ASPR/BARDA Pharmaceutical Countermeasures Infrastructure (PCI) Contracts Management and Acquisition (CMA) O’Neill Federal Office Building Washington, DC 20515 Email: [**] 5. No additional funding is incorporated into the Contract under this modification. The USG cost share obligation of $[**] remain unchanged. The Contract ceiling amount of $225,117,049.75, which includes the Contractor cost share amount, remains unchanged as follows:

Contract No. HHSO100201200004I Modification No. 0030 Page 4 of 3 [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] [**] Total Budget (Funded) $ [**] [**] *] [**]] [**] [**] [**] [**] $ 225,117,049.75 a) Any work performed by the Contractor beyond the funding or costs limit will be at the contractor’s risk. b) Cost reporting requirements are subject to FAR 52.232-20 Limitation of Cost (Apr 1984). All other terms and conditions remain unchanged and in full effect.
EXHIBIT 31.1
CERTIFICATION
I, Robert G. Kramer, certify that:
(1) I have reviewed this Quarterly Report on Form 10-Q of Emergent BioSolutions Inc.;
(2) Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
(3) Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
(4) The registrant's other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the registrant's disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the registrant's internal control over financial reporting that occurred during the registrant's most recent fiscal quarter (the registrant's fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant's internal control over financial reporting; and
(5) The registrant's other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant's auditors and the audit committee of the registrant's board of directors (or persons performing the equivalent functions):
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant's ability to record, process, summarize and report financial information, and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant's internal control over financial reporting.
Date: November 5, 2021
/s/ROBERT G. KRAMER
Robert G. Kramer
Chief Executive Officer
EXHIBIT 31.2
CERTIFICATION
I, Richard S. Lindahl, certify that:
(1) I have reviewed this Quarterly Report on Form 10-Q of Emergent BioSolutions Inc.;
(2) Based on my knowledge, this report does not contain any untrue statement of a material fact or omit to state a material fact necessary to make the statements made, in light of the circumstances under which such statements were made, not misleading with respect to the period covered by this report;
(3) Based on my knowledge, the financial statements, and other financial information included in this report, fairly present in all material respects the financial condition, results of operations and cash flows of the registrant as of, and for, the periods presented in this report;
(4) The registrant's other certifying officer and I are responsible for establishing and maintaining disclosure controls and procedures (as defined in Exchange Act Rules 13a-15(e) and 15d-15(e)) and internal control over financial reporting (as defined in Exchange Act Rules 13a-15(f) and 15d-15(f)) for the registrant and have:
(a) Designed such disclosure controls and procedures, or caused such disclosure controls and procedures to be designed under our supervision, to ensure that material information relating to the registrant, including its consolidated subsidiaries, is made known to us by others within those entities, particularly during the period in which this report is being prepared;
(b) Designed such internal control over financial reporting, or caused such internal control over financial reporting to be designed under our supervision, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles;
(c) Evaluated the effectiveness of the registrant's disclosure controls and procedures and presented in this report our conclusions about the effectiveness of the disclosure controls and procedures, as of the end of the period covered by this report based on such evaluation; and
(d) Disclosed in this report any change in the registrant's internal control over financial reporting that occurred during the registrant's most recent fiscal quarter (the registrant's fourth fiscal quarter in the case of an annual report) that has materially affected, or is reasonably likely to materially affect, the registrant's internal control over financial reporting; and
(5) The registrant's other certifying officer and I have disclosed, based on our most recent evaluation of internal control over financial reporting, to the registrant's auditors and the audit committee of the registrant's board of directors (or persons performing the equivalent functions):
(a) All significant deficiencies and material weaknesses in the design or operation of internal control over financial reporting which are reasonably likely to adversely affect the registrant's ability to record, process, summarize and report financial information, and
(b) Any fraud, whether or not material, that involves management or other employees who have a significant role in the registrant's internal control over financial reporting.
Date: November 5, 2021
/s/RICHARD S. LINDAHL
Richard S. Lindahl
Chief Financial Officer
EXHIBIT 32.1
CERTIFICATION PURSUANT TO 18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Quarterly Report on Form 10-Q of Emergent BioSolutions Inc. (the "Company") for the period ended September 30, 2021 as filed with the Securities and Exchange Commission on the date hereof (the "Report"), the undersigned, Robert G. Kramer, Chief Executive Officer of the Company, hereby certifies, pursuant to 18 U.S.C. Section 1350, that:
(1) The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
(2) The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
Date: November 5, 2021
/s/ROBERT G. KRAMER
Robert G. Kramer
Chief Executive Officer
EXHIBIT 32.2
CERTIFICATION PURSUANT TO 18 U.S.C. SECTION 1350, AS ADOPTED PURSUANT TO
SECTION 906 OF THE SARBANES-OXLEY ACT OF 2002
In connection with the Quarterly Report on Form 10-Q of Emergent BioSolutions Inc. (the "Company") for the period ended September 30, 2021 as filed with the Securities and Exchange Commission on the date hereof (the "Report"), the undersigned, Richard S. Lindahl, Chief Financial Officer of the Company, hereby certifies, pursuant to 18 U.S.C. Section 1350, that:
(1) The Report fully complies with the requirements of Section 13(a) or 15(d) of the Securities Exchange Act of 1934; and
(2) The information contained in the Report fairly presents, in all material respects, the financial condition and results of operations of the Company.
Date: November 5, 2021
/s/RICHARD S. LINDAHL
Richard S. Lindahl
Chief Financial Officer